An adolescent male presenting with multiple congenital limb defects planned for corrective surgery was incidentally detected to have low oxygen saturation on room air at rest. The child was not on any medication. After a pediatric cardiologist opinion a 2D Echocardiography and ECG were done which turned out to be normal. Proper history, detailed examination and simple bedside test carried out by the pediatricians proposed the possibility of methemoglobinemia. Since the child was not on any medication and after ruling out the possibility of toxin intake, a clinical diagnosis of congenital methemoglobinemia was arrived at, which was confirmed by the blood investigations. Since, systemic methemoglobinemia is a rare cause of cyanosis and has a varied presentation depending on the type and cause, a high index of suspicion is needed for the diagnosis. The coexistence of multiple congenital limb defects prompts one to think about a possible co-relation.
Methemoglobinemia, Congenital, Clubfoot, Brachydactyly, Vitamin C, Ascorbic acid
Methemoglobinemia (metHb) is a rare cause of cyanosis which does not improve on administering oxygen. Its congenital variant is an even rarer cause [1]. Some amount of MetHb is normally formed due to exposure to oxidative free radicals within red blood cells (RBCs) and also if temporarily donated electron is not recaptured by iron atom during unloading of O2 at tissue level, a process called as auto-oxidation. Daily around 3% Hb is converted to MetHb. However, the protective enzyme reducing system present in RBCs called as Nicotinamide adenine dinucleotide (NADH) dependent cytochrome b5 reductase converts 99% metHb back to HbA, so that the normal levels of metHb in blood is only 1-2%. There is also a minor alternative pathway that converts metHb to HbA comprising of glucose 6 phosphate dehydrogenase (G6PD) enzyme system [2].
Methemoglobinemia is a condition caused due to increased levels of methemoglobin levels in blood. Here we present a rare case of Congenital Methemoglobinemia discovered during workup for other condition.
An eleven-year-old boy, presented to orthopaedic outpatient department for deformity in right foot since birth and pain in both lower limbs off and on for past 1 year. Pain was more in the right lower limb, dull aching and aggravated on excessive walking and playing. On asking leading questions, mother also gave history that the child had easy fatigability as compared to his other siblings and peer group. There was also history of off and on bluish discoloration of lips and fingertips. There was no history of recent worsening of symptoms, dyspnea on exertion, palpitations, history suggestive of cyanotic spells, recurrent lower respiratory tract infections, hemoptysis. There was also no history of recent or chronic drug intake. Child was a 6th product of non-consanguineous marriage, born at term with immediate cry and an uneventful perinatal course. Child was developmentally normal and was studying in class fifth. First sibling had died at 3 months of age due to meningitis. All other siblings were apparently asymptomatic.
On examination child had slate gray complexion. Pulse rate was 88 per minute, respiratory rate was 22 per minute and pulses and blood pressure in all four limbs were normal. Mild peripheral and central cyanosis was present (Figure 1). Grade 3 clubbing was also present and saturation was 88% on room air. Multiple limb defects in upper limbs in the form of brachydactyly (bulbous distal end of 2nd finger of right hand and shortening of 3rd finger of both hands) and lower limbs in the form of congenital clubfoot on right side and shortening of 2nd and 4th digit of left foot were also noted (Figure 2). Systemic examination was within normal limits.
 Figure 1: Mild peripheral cyanosis. View Figure 1
Figure 1: Mild peripheral cyanosis. View Figure 1
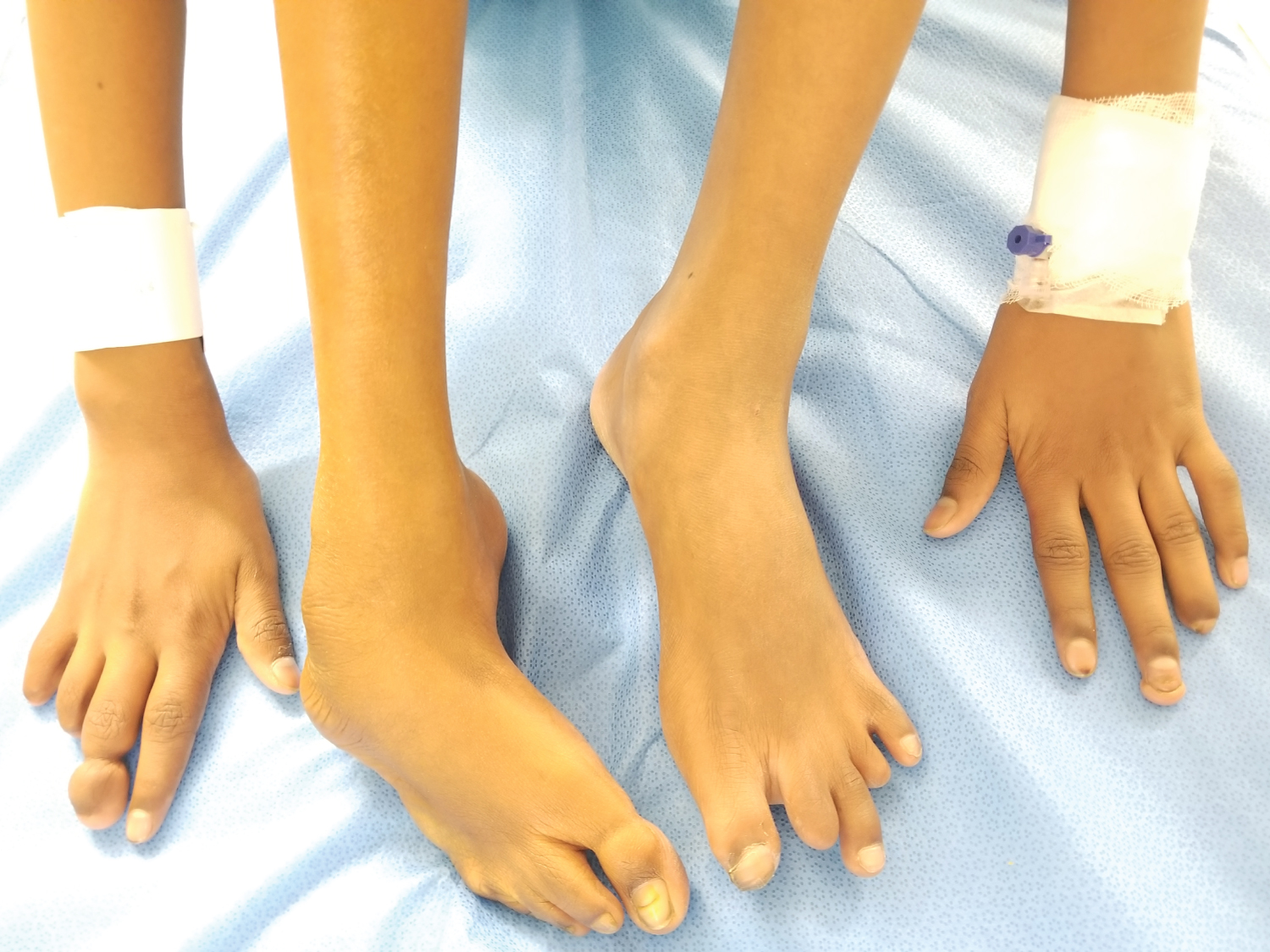 Figure 2: Congenital limb deformities. View Figure 2
Figure 2: Congenital limb deformities. View Figure 2
Blood investigations revealed hemoglobin of 13.5 g/dl with red blood count of 4.5 × 106 cells/mm3, total leucocyte count of 6500 cells/mm3, and platelet count of 2 × 105 cells/mm3 packed cell volume (PCV) - 36%, and mean corpuscular volume - 79.3 fl. X-Ray Chest was normal. 2D-Echo with Doppler study done by pediatric cardiologist was also normal. Blood drawn blood gas analysis was chocolate brown in colour (rapid screening filter paper test) and it showed pH - 7.43, PaCO2 - 33.2 mmHg, PaO2 - 45.9 mmHg, HCO3 - 22.5 mmol/L, and SO2% - 82.1% (Figure 3). Co-oximetry showed elevated methemoglobin level of 28.9% which clinched the diagnosis (Figure 4). Since there were no acute symptoms or any history of drug intake, congenital methemoglobinemia was suspected and further investigations were done. High-performance liquid chromatography (HPLC) was within normal limits, so were G6PD levels (9.30 U/g Hb; normal lab reference 4.6 to 13.5 U/g Hb). Cytochrome b5 reductase levels could not be assessed due to the non availability of the test in routine labs. Since there were no neurological symptoms and child was apparently asymptomatic a diagnosis of congenital methemoglobinemia type 1 was made. X-Ray of hands showed absent distal phalanx of right 2nd and 3rd and absent middle phalanx of right 5th finger, hypoplastic distal phalanx of left 4th finger. X-ray of left foot showed absent middle phalanx of 2nd and 4th toe. Right foot X-ray showed changes in bones due to prolonged walking on untreated clubfoot without absence of any bone (Figures 5).
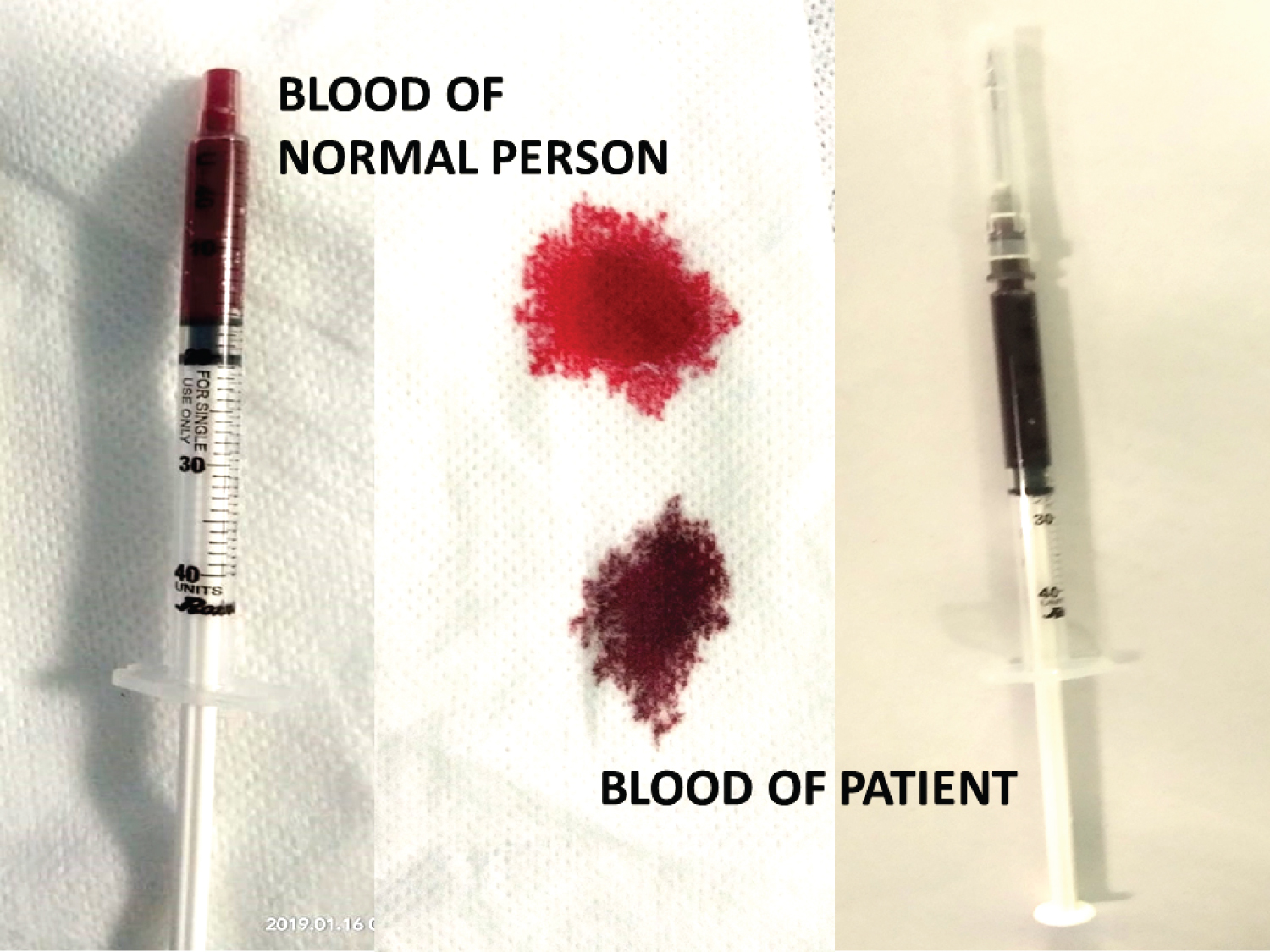 Figure 3: Rapid Screening Test. View Figure 3
Figure 3: Rapid Screening Test. View Figure 3
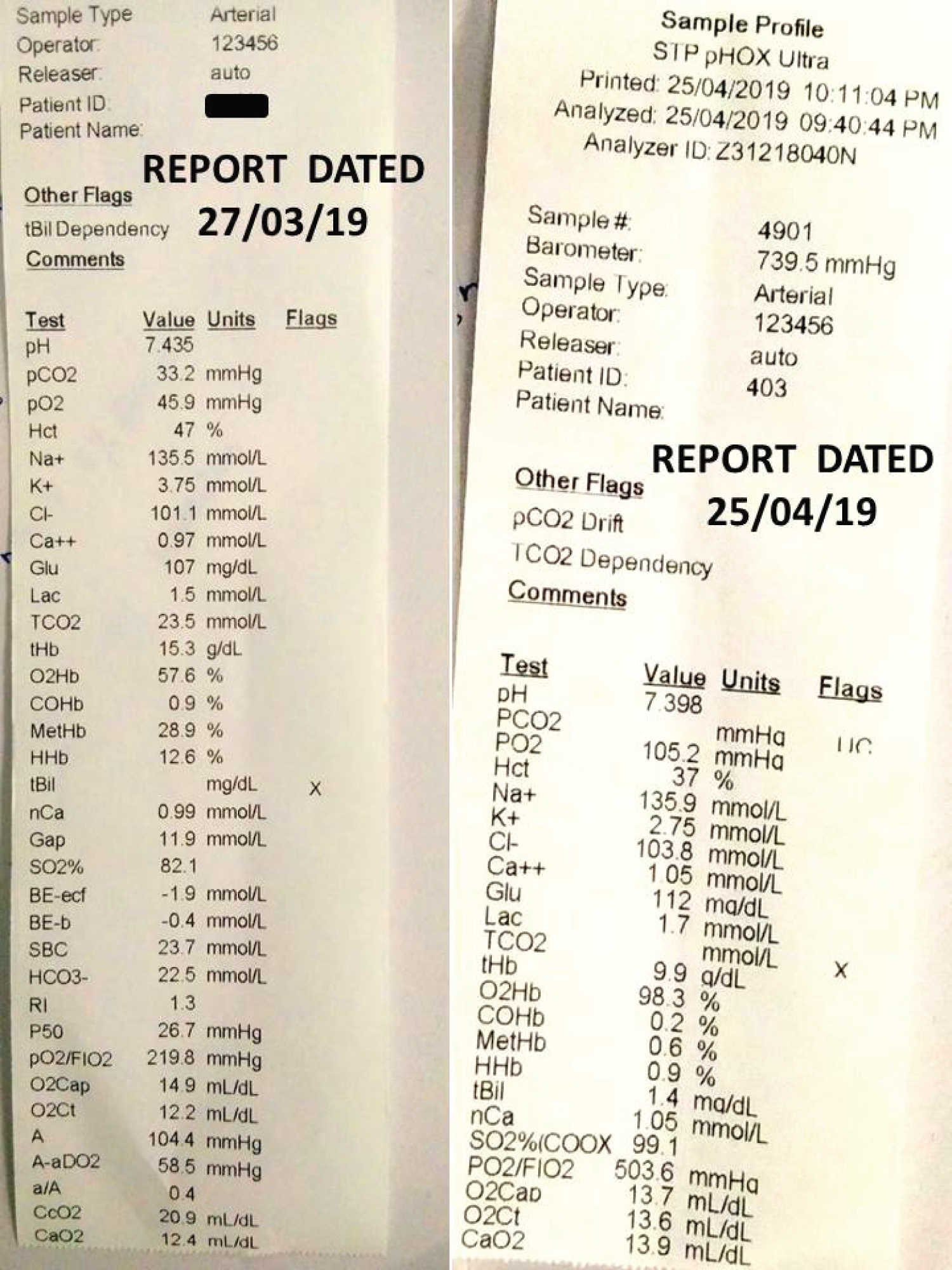 Figure 4: Blood Gas Analysis on (a) 27/03/2019; (b) 25/04/2019. View Figure 4
Figure 4: Blood Gas Analysis on (a) 27/03/2019; (b) 25/04/2019. View Figure 4
Initially the surgery for CTEV was deferred by anesthesiologist for low saturation. Child was started on vitamin C 250 mg twice daily. Follow up after 1 month showed reduction in methemoglobin levels to 0.6% and saturation improved to 95%- 99% on room air (Figure 4). Care was taken to avoid any oxidative drug and also ascorbic acid and methylene blue injections were kept on standby. One unit packed cells was also arranged. Patient was operated for CTEV without any perioperative complications (Figure 5). Further genetic work up for congenital methemoglobinemia as well as brachydactyly was desirable but could not be done due to financial constraints (Figure 6). Patient is under follow up for past 1 year and regularly taking Vitamin C tablets with sustained improvement in SpO2 levels. There has been no relapse of deformity corrected (Figure 7).
 Figure 5: X-rays of (a) Hands and (b) Feet of the child. View Figure 5
Figure 5: X-rays of (a) Hands and (b) Feet of the child. View Figure 5
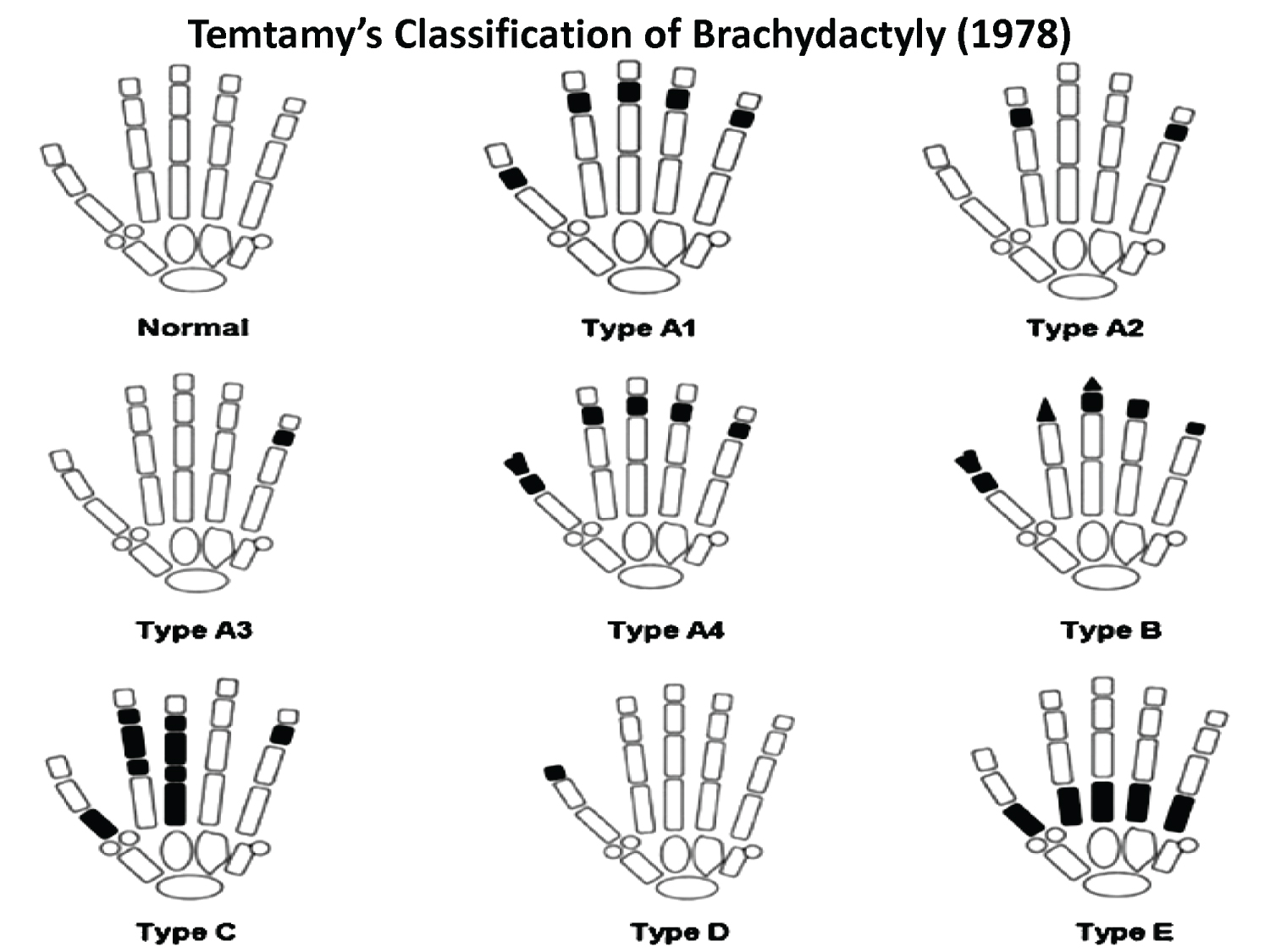 Figure 6: Types of Brachydactyly (Reproduced from Birth Defects: Original Article Series (Genetics of Hand Malformations - Brachydactyly; Samia A. Temtamy & Victor A. McKusick) (1978)). View Figure 6
Figure 6: Types of Brachydactyly (Reproduced from Birth Defects: Original Article Series (Genetics of Hand Malformations - Brachydactyly; Samia A. Temtamy & Victor A. McKusick) (1978)). View Figure 6
 Figure 7: Pre-correction and Post-correction follow up figure of Clubfoot Deformity. View Figure 7
Figure 7: Pre-correction and Post-correction follow up figure of Clubfoot Deformity. View Figure 7
The adult hemoglobin (HbA) consists of four folded polypeptide chains (two alpha and two beta), each of which has porphyrin heme ring attached. The centre of each heme ring is an atom of iron in Ferrous (Fe2+) state. These functional centres of hemoglobin (Hb) molecule bind reversibly to oxygen (O2) by temporary partial transfer of electron (oxidation) from Fe2+ to O2 to form oxyhemoglobin (oxyHb, superoxo ferriheme, Fe3+O2-) when partial pressure of oxygen is high (as in lungs). When partial pressure of O2 is low as in peripheral tissues, the oxygen is unloaded from oxyHb recapture of temporarily shared electron by iron atom, thus, returning to ferrous state [3]. Methemoglobin (MetHb) is a form of Hemoglobin (Hb) which contains ferric (Fe3+) form of iron instead of the ferrous (Fe2+) form. The affinity for oxygen of ferric iron is impaired as it cannot donate electron for binding of O2.
Methemoglobinemia can be acquired or congenital. Acquired metHb is due to oxidative drugs when given to susceptible individuals i.e. with G6PD deficiency. Classical drugs that cause metHb include various drugs (trimethoprim, sulfonamides, metoclopramide, dapsone), local anesthetics (especially articaine, benzocaine, prilocaine, lidocaine), and toxins (aniline dyes, nitrates, chlorates, and bromates) [4]. Congenital metHb is due to NADH dependent cytochrome b5 metHb reductase enzyme deficiency. It is of two types - Type I, when the deficiency is restricted to RBCs and Type II, when the deficiency is global including cells in brain. Type II is extremely rare, associated with mental retardation and neurological abnormalities that result in death during childhood. Type I is associated with normal life expectancy and presents as uncomplicated, benign methemoglobinemia with occasional dyspnea and fatigability. Type I is usually detected later in childhood or adulthood, mostly accidently as the cases are asymptomatic, especially in early period [5]. In metHb, the erythrocytes are unable to transport sufficient amount of oxygen to the body tissues. This shifts the oxygen dissociation curve to the left, thus lowering the oxygen release of hemoglobin. The blood is chocolate brown in color. The low oxygen concentration in blood leads to cyanosis. The presence of symptoms in an individual depends on the percentage of MetHb in blood. At metHb levels between 3%-15%, there is greyish blue discoloration of the skin; at 15%-30%, chocolate-brown blood is visible; at levels between 30%-50% headache, dyspnea, lightheadedness, weakness, confusion, palpitations and chest pain may occur, at levels between 50%-70% cardiac arrhythmias, delirium, seizures, coma develop and the condition is usually fatal above MetHb levels of > 70% [6,7].
Congenital methemoglobinemia is inherited as a autosomal recessive disease due to mutation in CYB5R3 gene resulting in deficiency of NADH dependent Cytochrome B5 MetHb reductase levels within cells [8,9]. A minor variant of congenital metHb exists where there is hemoglobin M (HbM), with substitution of amino acid in either the alpha or beta part of the hemoglobin. These individuals may also develop methemoglobinemia due to decreased ability of the enzyme to reduce iron back to its Fe2+ state [10,11].
In congenital methemoglobinemia, the patient may either be asymptomatic or appear to have cyanosis. Till date only 26 cases of congenital methemoglobinemia (including this case) have been reported in English literature. Nineteen of them were diagnosed as Type 1 and 6 as Type 2 [12]. The median age of diagnosis of type 1 methemoglobinemia was 31 years with cyanosis and shortness of breath being the most common sign and symptom. Since most of the patients with Type 1 methemoglobinemia are asymptomatic, these cases may be underdiagnosed or incidentally diagnosed as in our case. However, they are still prone to life-threatening acute episodes which can be prevented by the early diagnosis and treatment. No definitive treatment is available, but vitamin C, due to its antioxidant properties has been shown to reduce the levels of methemoglobin and thus improve the oxygen saturation [13]. Other therapeutic option is methylene blue injection for acute episodes.
Our patient also had associated limb defects in the form of brachydactyly and club foot. Brachydactyly refers to condition in which there are disproportionately short fingers and toes. It is due to congenital absence or underdevelopment of metacarpal or phalangeal bones. The condition may be isolated or associated with other anomalies and syndromes. The isolated form is a usually an inherited in an autosomal dominant pattern. It is classified in various patterns, the most common classification being in which brachydactyly is grouped into five groups (A-E) [14]. We could not fit our patient in any one particular group according the classification nor we could find an autosomal dominant pattern of inheritance of brachydactyly (Figure 6). On extensive literature search, no association was found between congenital methemoglobinemia and brachydactyly. However, the limb anomalies were significant and mutation analysis could have revealed some genetic defect, hence is warranted in this case. But due to financial constraints we were not able to send the further genetic work up.
Little is known about the exact incidence and prevalence of congenital methemoglobinemia [15]. With more and more cases reported, most of which are detected incidentally, the prevalence ought to be higher than thought. We also endorse the formation of a national level registry for congenital methemoglobinemia [16]. This will further help to unveil the geographical distribution of the disease, its prevention by genetic counseling, development of newer therapeutic strategies and in genetic research.
Methemoglobinemia is a rare cause of cyanosis that does not improve with administration of O2. Low O2 saturation at rest and typical brown colour of blood should raise suspicion about the diagnosis. Vitamin C (ascorbic acid) and Vitamin B2 (riboflavin) help in reducing the levels of methemoglobin in congenital variant. Methylene Blue is useful in acute acquired methemoglobinemia. A high index of suspicion is required to diagnose the cases of congenital metHb, especially as a part of preanaesthesia screening for invasive procedures. Co-relation with limb congenital defects needs to be established with further genetic studies.
Sheetal Agarwal: Diagnosis, workup, management of pediatric aspect of the case/condition, manuscript editing.
Dhirendra Singh: Diagnosis, workup, manuscript inputs.
Ankur Agarwal: Management of orthopaedic aspect of the case, patient followup, manuscript writing, manuscript revision, correspondence.
Ridhima Sharma: Anaesthetic concerns of the case, manuscript editing.
Savitri Singh: Workup, pathological aspect of the case.
We would like to thank the dedicated team of Anaesthesiologists - Dr. Mukul Jain, Dr. Poonam Motiani and Dr. Vibha Chhabra for their untiring and ever zealous efforts.
None.
None.