A 61-year-old woman was admitted to our ward - after her third visit to the Emergency Department in two months - because of progressive lethargy, weakness, and persistent euvolaemic hyponatraemia. Based on the results of the first diagnostic workup (complete blood count, serum and urinary biochemistry), a diagnosis of Syndrome of Inappropriate Antidiuretic Hormone Secretion (SIADH) was established. A comprehensive second diagnostic workup was performed, leading to the diagnosis of anterior hypopituitarism due to primary empty sella syndrome, a rare multiple hormone deficit which could result in devastating consequences if left untreated.
Hyponatraemia, Syndrome of inappropriate antidiuretic hormone secretion, Adrenal insufficiency, Hypopituitarism, Empty sella
CT: Computed Tomography; ADH: Antidiuretic Hormone; SIADH: Syndrome of Inappropriate Antidiuretic Hormone Secretion; BID: bis in die (twice a day); ACTH: Adrenocorticotropic Hormone; QD: quaque die (once a day); GH: Growth Hormone; IGF-1: Insulin-like Growth Factor 1; MRI: Magnetic Resonance Imaging; PES: Primary Empty Sella; PESS: Primary Empty Sella Syndrome; CRH: Corticotropin-Releasing Hormone
Hyponatraemia is the most common hydro-electrolytic derangement in clinical practice, often overlooked. We present the case of a 61-year-old woman with persistent euvolaemic hyponatraemia, initially labeled as a syndrome of inappropriate antidiuretic hormone (ADH) secretion (SIADH). The proper workup led to the diagnosis of a rare multiple hormone deficit, with devastating consequences for the patient if left untreated. Our case highlights the importance of a comprehensive approach to the patient with euvolaemic hyponatraemia.
A 61-year-old woman with no significant past medical history was admitted to our ward because of persistent hyponatraemia. Approximately one year before, she was diagnosed with mixed anxiety-depressive disorder, characterized by hyporexia, weight loss, nausea, asthenia, and apathy. A blood chemistry test showed no relevant findings. She started treatment with amitriptyline and saw moderate improvement. Two months prior to the admission, she came to the Emergency Department twice in five days complaining of difficulty urinating, weakness, dry mouth, blurry vision, and tremors. On both occasions, the biochemical evaluation showed severe hyponatraemia (Table 1, ED 1 and ED 2). However, it was only partially corrected with hypertonic saline solution. Amitriptyline was discontinued and no further investigation was carried out before discharge. The patient improved after discontinuing amitriptyline, but two weeks prior to most recent admission, she relapsed into her anxious-depressive state, with new onset of progressive somnolence, gait instability, nausea, anorexia and urinary incontinence, leading to an Emergency Department visit.
Table 1: Laboratory data. View Table 1
Upon admission, the patient was haemodinamically stable and afebrile. Intense somnolence and bradyphrenia without neurological focality was noted on the physical examination. There were no signs of hypovolaemia (hypotension, orthostatism, dry skin and mucous membranes) or hypervolaemia (jugular vein distention, oedema, ascites), so she was classified as euvolaemic. A brain CT scan was performed, showing no abnormal findings. Blood and urine chemistry tests (Table 1, day 0) documented severe hyponatraemia with serum sodium 113 mmol/L, plasma hyposmolality, inappropriate urine concentration, and low urinary sodium (attributed to a low oral uptake of salt). The moderate-severe hyponatraemic encephalopathy was initially treated with hypertonic saline solution (3% NaCl). Sodium levels rose 9 mmol/L in the first 24 hours (Table 1, day +1) with mild clinical improvement. Following diagnosis of persistent euvolaemic hyponatraemia compatible with SIADH, the patient was transferred to our ward for a second diagnostic workup. Fluid restriction (1000 mL/day) and oral loop diuretics (furosemide 40 mg BID) were started, resulting in a progressive, controlled increase of serum sodium concentration (Table 1, days +2 and +4). A CT scan of the chest and abdomen was performed in order to rule out occult neoplasia; it showed no relevant findings. Serum hormone assays were also requested, revealing hypofunction of the thyroid axis and undetectable levels of cortisol and adrenocorticotropic hormone (ACTH) (Table 2). Reviewing the electronic medical records, we found mild hypoglycaemia (about 65 mg/dL) in several previous biochemical tests, along with occasional hypotension (about 80-90/45-50 mmHg).
Table 2: Results of the hormone assay. View Table 2
Repeated hormone assays confirmed the previously-found deficits, and also revealed low levels of gonadotropins, growth hormone (GH) and insulin-like growth factor 1 (IGF-1) (Table 2). A diagnosis of anterior hypopituitarism was established, and replacement therapy with oral hydrocortisone (20 mg BID) and levothyroxine (50 μg QD) was started. Fluid restriction and furosemide were discontinued. The patient reported she was nulligravida, had normal menstruations until she was 55-years-old, denied galactorrhoea and polyuria, and had never taken corticosteroids. No previous cranioencephalic trauma, major bleeding, surgical interventions, or radiation therapy were reported. A pituitary MRI scan was performed, showing an empty sella (Figure 1).
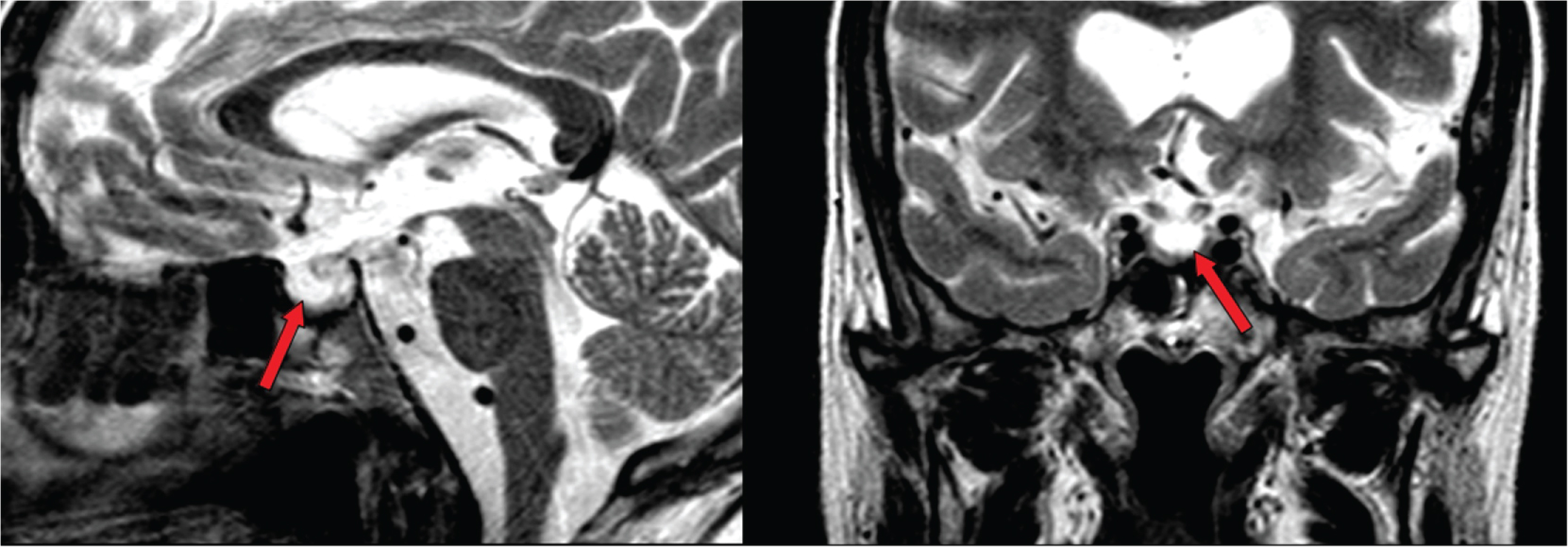 Figure 1: Sagittal (left) and coronal (right) T2 sections from the MRI scan, showing an empty sella filled with cerebrospinal fluid (arrows).
View Figure 1
Figure 1: Sagittal (left) and coronal (right) T2 sections from the MRI scan, showing an empty sella filled with cerebrospinal fluid (arrows).
View Figure 1
The patient's clinical condition improved over the following days, and serum sodium concentration, blood pressure, and blood glucose levels normalized. She was discharged with oral hydrocortisone 10-5-5 mg and levothyroxine 50 μg QD. One month later, another biochemical and hormone evaluation was performed (Table 1, month +1), which showed eunatraemia and persistence of the hormone deficits. One year after discharge she remained stable, and her physical and mental condition recovered completely.
Anterior hypopituitarism secondary to primary empty sella syndrome (PESS). Severe euvolaemic hyponatraemia due to secondary adrenal insufficiency.
Empty sella is characterized by the herniation of the subarachnoid space within the sella and the flattening of the hypophysis. It is classified as primary (PES) in cases in which aetiology is unknown after excluding a history of previous pituitary pathological conditions, such as radiation therapy or surgery. PES is a relatively common incidental finding in autopsy and radiological imaging (8-35% in the general population), and is asymptomatic in the majority of cases [1]. When PES is associated with symptoms, it is called primary empty sella syndrome (PESS). Though endocrine abnormalities are documented in around 19% of patients, panhypopituitarism is infrequent [1].
A prospective study which included 573 patients with euvolaemic hyponatraemia who met the criteria for SIADH reported that 1.5% was actually found to have secondary adrenal insufficiency due to hypopituitarism [2]. In the secondary adrenal insufficiency, ADH and corticotropin-releasing hormone (CRH) are secreted together from the hypothalamus due to a lack of negative feedback from ACTH and cortisol [3]. Clinically and biochemically, it is almost impossible to distinguish it from SIADH, but it can be suspected if there are signs or symptoms of hypocortisolism (hypotension, hypoglycaemia) or other hormone deficits. The diagnosis is confirmed by assessing adrenal axis function (serum cortisol and ACTH levels, ACTH stimulation test, if necessary) [3,4].
Patients with euvolaemic hyponatraemia who met the criteria for SIADH with no obvious aetiology should be studied thoroughly. Medications are a frequent cause (especially antidepressants, antipsychotics, and anticonvulsants) and any candidate drug should be suspended if possible. If hyponatraemia persists or if the patient does not take these types of drugs, an additional diagnostic workup is warranted. It is recommended to request, at minimum, a chest X-ray and a complete biochemical evaluation with hormone assays for the evaluation of the adrenal and thyroid axes. If no aetiology has still been identified or if there is a strong clinical suspicion of neoplasia, then CT scans of the skull, chest and abdomen should be performed [5].
The authors declare no conflicts of interest. No external financial support was received.
All authors contributed equally to the manuscript.