Case report and literature review.
To document the case of a type II segmental spinal dysgenesis and to provide a literature review that describes the key elements of this rare entity.
The segmental spinal dysgenesis (SSD) is a rare and complex spinal dysraphism. We document a case of a type II SSD in a full-term female neonate with her clinical symptoms, imaging characteristics and intraoperative findings, depicting the rare association with a pure meningocele and its primary management. In addition, we performed a literature review, with special emphasis on the embryological and radiological classification, aiming to a proper diagnostic and therapeutic approach of this entity.
Spinal dysgenesis, Spinal dysraphism, Meningocele
MRI: Magnetic resonance imaging; CT: Computed tomography scan
Segmental spinal dysgenesis is a rare and complex entity characterized by a dysgenesis of the thoracolumbar or lumbar spinal segment, usually associated with congenital scoliosis, focal abnormalities of the spinal cord and the spinal nerves. Taking into account the embryological pathway, this spinal dysraphism can be further subdivided into two main types that present different imaging findings, as well as different therapeutic requirements. Here we present the case of a new born female patient with a type II SSD associated with a protruding meningocele.
Full-term female-neonate, delivered by cesarean section without complications, with prenatal ultrasound documentation of bilateral congenital talipes equinovarus and suggestive imaginological signs of a non-specific thoracolumbar spine malformation. At the time of birth, the physical examination reflected the presence of a dorsal complex cutaneal appendix covered by skin, associated with lower limbs hypertonia and bilateral talipes equinovarus. There were no respiratory alterations. MRI images of the neuroaxis were performed, identifying an abrupt interruption of the thoracolumbar spine with a dysmorphic block vertebrae with spondyloptosis, posterior closure defects of several vertebral segments and significant spinal canal stenosis associated with spinal cord narrowing at the apex of the retrolisthesis, with bulky and low lying cord at the lumbar segments. The vertebral anomalies were better characterized by 3D CT reconstruction, confirming the findings previously described (Figure 1) with a significant compromise of the bony elements of the thoracolumbar junction.
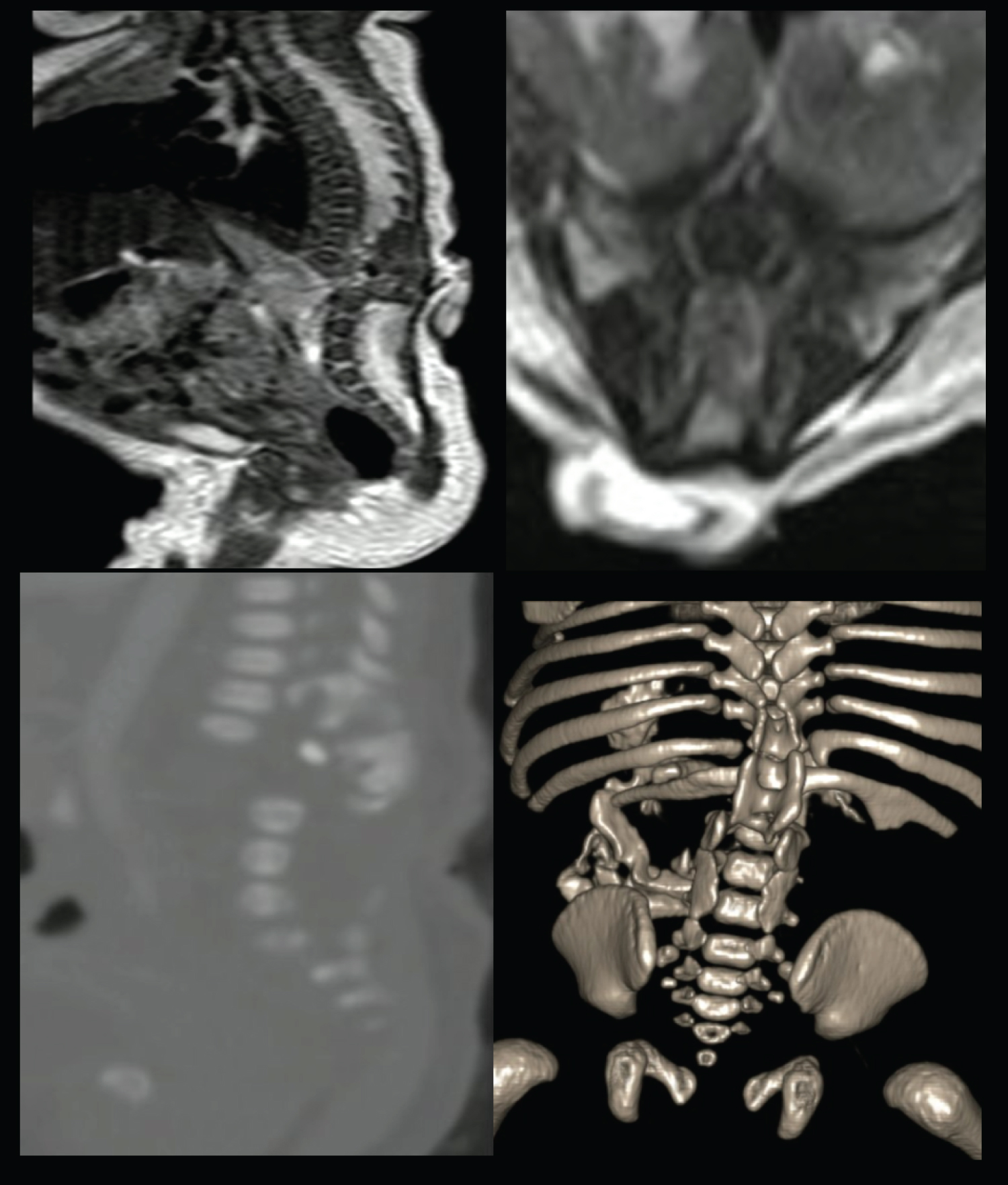 Figure 1: Sagittal and axial MRI of the spine identifying an abrupt interruption of the thoracolumbar spine with an associated dysmorphic block vertebrae. Spine CT and 3D reconstructions depict the vertebral anomalies compatible with segmental spinal dysgenesis. View Figure 1
Figure 1: Sagittal and axial MRI of the spine identifying an abrupt interruption of the thoracolumbar spine with an associated dysmorphic block vertebrae. Spine CT and 3D reconstructions depict the vertebral anomalies compatible with segmental spinal dysgenesis. View Figure 1
The patient underwent surgery for the appendix excision identifying a meningocele with primary closure without complications (Figure 2), surgery that was performed one week after being born. The extremities abnormalities as well as the spine deformity are going to be surgically intervened when the patient is at least six-months-old. At the last follow up, one month after surgery, there was neurological stability.
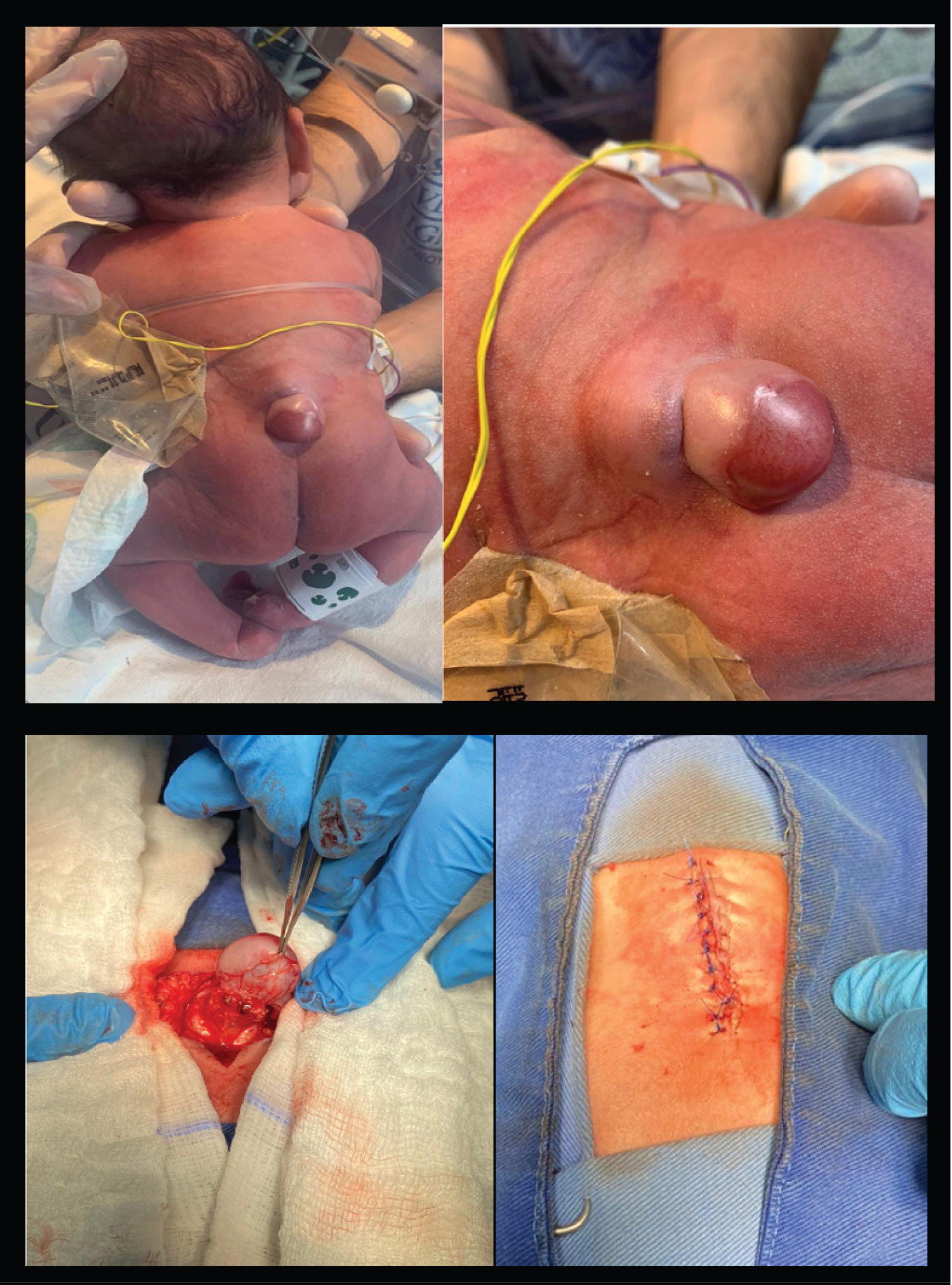 Figure 2: Complex cutaneous appendix that was correlated with a meningocele.
View Figure 2
Figure 2: Complex cutaneous appendix that was correlated with a meningocele.
View Figure 2
Segmental spinal dysgenesis is a rare and complex entity characterized by a dysgenesis of the thoracolumbar or lumbar spinal segments, usually associated with congenital coliosis, focal abnormalities of the spinal cord and the spinal nerves [1], recognized by Scott, et al; in 1988 [2-4]. There is no sex predilection, and associations with maternal diabetes, anoxia and intrauterine toxins exposure have been established [1,5]. Consistently, There is a superior spinal cord with normal appearance, continued by an abnormally thin spinal cord with no nervous roots, and an inferior low lying and bulky distal spinal cord segment [1,2].
Based on the imagenological and clinical findings, several authors [4,6,7] have defined this condition as an uncommon and complex occult spinal dysraphism that meet certain criteria: Congenital paraparesis or paraplegia with lower limb abnormalities, multiple anomalies in the vertebral formation or segmentation with associated scoliosis, absence or segmental malformation of the spinal cord and the spinal nerves and identification of a spinal cord segment distal to the interrupted cord [5,7]. It has been further described an associated local stenosis and deformity of the spinal canal with a bone ring surrounding the cord segment at the level of the dysgenesis, posterior displacement of the affected vertebral bodies that present central junction defects, pedicle alterations as well as malformations of the spinous and transverse processes and nerve roots irregularities at the level of the stenosis [8,9]. Concomitantly, it is possible to identify visceral associated abnormalities such as horseshoe kidneys, dextrocardia and joint compromise (congenital hip displasia) [5,8].
Several equivalent terms have been described to name this entity: Congenital vertebral displacement, congenital dislocation of the spine, congenital duplication of the spinal canal, thoracolumbar spondyloptosis, among others [8], without there being a single recognized terminology for this condition and neither a standard course of treatment.
Within a clinical point of view, the most frequent reported presentation is spastic paraparesis associated with neurogenic bladder [1,5], however, the clinical picture may vary depending on the level as well as the extent of the malformation, the kyphosis degree, and others systemic abnormalities [10,11]. Although some neonates are neurologically unaffected at birth, the motor function alteration tends to be progressive, and if it is left untreated, it may evolve to paraplegia [12] given the presence of progressive instability of the spine [8,10].
Magnetic resonance imaging is the modality of choice for a diagnostic approach [1], although its presence may be suggested in prenatal ultrasound by the detection of altered vertebral segmentation associated with lower limb malformations [13]. In addition to the previously described imaging features, this entity may course with an associated occult spinal dysraphism such as tethered cord or lipoma of the filum terminale [1,5,10]. On the other hand, the primary role of the computed tomography lies in the surgical planning [6].
Based on the embryological and imaging characteristics, this entity can be divided into two types [5]:
Type I: Defined as a congenital segmental abscence/dysgenesis of the vertebral bodies, spinal cord or nerve roots without retrospinal protrusion, gibbus deformity or spinal stenosis, that occurs secondary to a gastrulation alteration of the chorda-mesoderm, were failures in its positioning leads to apoptosis of the poorly located area, related to hypoplasic or absent spinal cord levels. This anomaly compromises the notochordal function and somite induction leading to a developmental impairment of the spinal cord, spine and nerve roots [5].
Taking into account all of the above, the imaging characteristics of the type I segmental spinal dysgenesis are given by the presence of mild kyphoscoliosis without spinal canal stenosis and focal dysplasia or aplasia of the spinal cord that communicates the proximal spinal segment with a bulky and low lying spinal cord segment, that results from caudal cells addition. As there is a sclerotome involvement without a dermatome compromise, there is no open dysraphism coexistence [5-7].
Type II: A segmental abscence/dysgenesis of the vertebral bodies, spinal cord or nerve roots in association with severe canal stenosis (stenosis greater that 50% of the diameter) and severe kyphoscoliosis with significant gibbus deformity, as a result of a somatogenesis dysregulation that leads to a secondary spinal cord dysplasia by mechanical compression conditioned by a spondyloptosis, chronic Ischemia and a final state of cord hypoplasia at the level of the dysplasic vertebral bodies. Additionally, the distal segment of the spinal cord is thickened and has a lower insertion [5,7].
The appropriate imaging differentiation between the sub-types acquires special relevance when evaluating the therapeutic approach, since the embryological pathway of type I SSD does not respond to surgical management [5]. Taking into account the few cases reported in the literature, the treatment recommendations for this entity are not fully clarified, especially regarding the optimal time of intervention, as well as the techniques for spinal stabilization in those patients were the mechanical forces influence the spinal cord hypoplasia (type II SSD), that appears to present a significant clinical response to optimal surgical decompression [5]. Several reports have described sequential procedures before the age of two, recommending early fusion to avoid progressive neurological deterioration [9]. Some authors advocate a spinal fusión at the time of the diagnosis, however, treatment should be Individualized based on the imaging findings and patient clinical condition [11,14], recalling that the optimal treatment time, the use of orthesis, the extent of decompression nor instrumentation and type of approach are still in doubt [8,15,16].
The authors of this paper accept this submission and are responsible of its content, accepting its publication in this prestigious and respected journal.