Abelmoschus esculentus is widely cultivated and consumed across the globe for its nutritional and medicinal purpose. In spite of the growing demand, its cultivation is massively affected by various insects, fungi, nematodes and viruses. Due to lack of genomic and limited transcriptomic resources, genetic manipulation studies concerning the crop improvement against various environmental factors is scarce for this crop. Thereby, the present study aims to develop high quality transcriptome of A. esculentus by employing the Next-Generation based RNA sequencing of four cDNA libraries generated from the leaf samples. Sequencing yielded a total of 206.3 million paired-end clean reads with 66,382 assembled unigenes having a total length of 71.35 Mb, an average length of 1,074 bp and an N50 of 1,408 bp. About 56% of the unigenes were successfully annotated in four public databases including Pfam, GO, COG, and KEGG. GO analysis revealed that the majority of the annotated unigenes were involved in key biological processes like ATP binding, DNA binding, transcription, DNA-templated, and integral component of membrane. KEGG pathway analysis showed that 16,307 unigenes were assigned to 143 pathways in which majority of secondary metabolites related transcripts involving in phenylpropanoids, flavonoid and terpenoid biosynthesis pathway were identified. In addition, transcription factor and simple sequence repeats (SSRs) analyses revealed 76 transcription factor families and 9,578 potential SSRs in the A. esculentus leaf transcriptome. Furthermore, de novo assembled leaf transcriptome generated in the present study had longer transcripts with better N50 sizes and the quality of assembly was ensured by qRT-PCR analysis. The A. esculentus sequence information presented in this study will be a valuable resource for further molecular genetics and functional genomics studies for the improvement of this crop plant.
Abelmoschus esculentus, Bhendi, de novo, Assembly, Transcriptome
Abelmoschus esculentus (bhendi or okra) is an economically important food crop belongs to the family Malvaceae. It is grown all over the world mainly in the tropical and subtropical regions [1,2]. India ranks first in the world with 6.5 million tons (72% of the total world production) of bhendi produced from over 0.5-million-hectare land [3]. The edible premature green pod comprises the main source of dietary nutrients like calcium, iron, magnesium, manganese, vitamins A, B, C, K and folates [4]. It has also gained global recognition for pharmacological and medicinal properties against cancer, high-cholesterol, and diabetes mellitus [5-7].
Despite of global importance, its cultivation is affected by various insects, fungi, nematodes and viruses. In India, its cultivation is challenged by the occurrence of begomoviruses such as Yellow vein mosaic virus and Enation leaf curl virus (family: Geminiviridae), accounting for about 30-100% yield loss depending on the plant age and time of infection [8,9]. Biotechnological intervention to generate resistant cultivar against this viral infection is limited due to the lack of genome information. Till date, only very few sequences such as Chalone synthase mRNA, chloroplastic maturase K gene, partial sequences of ribosomes, NADH dehydrogenase, lycopene cyclase, and lycopene epsilon cyclase mRNA are available in the NCBI database from the entire genus. The first report of leaf and pod transcriptome of A. esculentus has been accomplished by Schafleitner, et al. [10]. However, the reported transcriptome is not ample enough to represent the whole transcriptome, owing to the low depth of sequencing as the assembled unigenes are smaller in sizes (mean length-309 nt) with low N50 of 321 bp [11]. Recently, we identified 128 known and 845 novel miRNAs from the leaf sample of A. esculentus along with the precursors by sRNA and precursor’s miRNA sequencing [12]. However, the respective complementary target genes for many of the known as well novel miRNAs could not be predicted by utilizing the reported transcriptome information in the database. Therefore, high-quality of sequence information on A. esculentus will be a valuable resource to understand gene expression by studying the miRNA-mRNA interactions, to generate resistant cultivar against various environment cues, to develop molecular markers for the cultivation of superior trait by plant breeders, and also to understand the various biological pathways for further genetic manipulation of this crop plant to improve its nutritional quality.
In the present study, we utilized Next Generation based-RNA sequencing (NGS) technology to obtain high quality leaf transcriptome of A. esculentus. A large number of transcript sequences involved in various biological processes were identified from the present transcriptome data. The present leaf transcriptome from A. esculentus had assembled transcripts with longer length, better mean length, and N50 size. The sequence information from the present study is available publicly and will facilitate better understanding of this crop plant for its future improvement.
Abelmoschus esculentus plants were grown in a controlled environment maintained with 16/8 h photoperiod at 25 ℃ in a plant growth chamber (Panasonic, Europe). Leaf samples from four healthy independent biological replicates were harvested, snap frozen in liquid N2 and stored at -80 ℃ till RNA extraction.
Total RNA was extracted from 100 mg of leaf samples using the modified Trizol method [12]. The extracted RNA samples were treated with DNase (Thermo Scientific, Waltham, MA, USA) at 37 ℃ for 30 min. DNase treated RNA from each sample was evaluated by Agilent Bioanalyzer 2100 (Agilent, Santa Clara, CA, USA) for its integrity. Only the RNA samples with RIN value more than 7.5 were used for the cDNA library preparation using Illumina Truseq RNA Sample Preparation Kit (Illumina Inc., San Diego, CA, USA).
For the cDNA library construction, 1 µg of total RNA was used to capture mRNA using oligo (dT) beads. Poly A captured mRNAs were fragmented for 2 mins at 94 ℃ in the presence of divalent cations and then reverse transcribed using Superscript III Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA) by priming with Random Hexamers. Second strand cDNA was synthesized using DNA Pol I and RNase H treatment followed by adapter ligation. Constructed cDNA library was amplified with 8 cycles for the enrichment of adapter-ligated fragments and subsequently paired-end sequencing was performed by Illumina NextSeq 500 (Illumina Inc., San Diego, CA, USA).
Raw reads with an average read length of 150 bp were processed using Trimmomatic program [13] for the removal of adapter sequences, low quality reads with phred score < 25, reads with ambiguous 'N' bases > 5% and read length below 36. Clean reads thus obtained were used for de novo assembly using Trinity (version 2.1.0) with k-mer size of 25. Resulting assembly statistics was evaluated using the program TransRate [14]. Transcripts redundancies were removed using the program CD-HIT with an identity parameter of 70% [15,16]. All the RNA sequencing raw reads generated from four cDNA libraries were deposited in NCBI Short Read Archive (SRA) under the accession numbers SRX2995608, SRX2995609, SRX2995611, and SRX2995612.
To derive the putative functions, assembled unigenes were subjected to BLASTX analysis against the UniProt protein database of green plants with an E-value cut-off ≤ 1e-05. BLASTX results were imported into BLAST2GO to assign unigenes with GO (gene ontology) terms describing biological process, molecular function and cellular component. Clusters of Orthologous Groups (COG) (https://www.ncbi.nlm.nih.gov/COG/) were performed based on the BLAST results by mapping unigenes against COG database. Further, unigenes were also assigned for the pathways interpretation using KEGG database [17]. From the KEGG analyses, unigenes corresponded to the secondary metabolites pathways (phenylpropanoids, flavonoid and terpenoid biosynthesis) were analysed using KEGG mapper (https://www.genome.jp/kegg/tool/map_pathway2.html).
To identify transcription factors in A. esculentus, all the assembled unigenes were searched against the plant transcription factor database using PlantTFcat [18].
Assembled unigenes from A. esculentus leaf transcriptome were searched for the detection of SSRs using the MISA tool (http://pgrc.ipk-gatersleben.de/misa/) with parameters as mentioned by Du, et al. [19].
In order to validate the assembly, eight transcripts (Histone lysine methyltransferase, Nudix hydrolase, Polycomb group embryonic flower 2 like isoform, Retinoblastoma related protein, Adenosine kinase, DNA directed RNA polymerase IV and V subunit 4, Dicer-like protein 4, and Disease resistant protein) were randomly selected and primers were designed with de novo assembled transcripts as the reference. DNase treated 1 µg of total RNA from leaves of A. esculentus was converted to cDNA according to the manufacturer's instructions provided with the RevertAid First Strand cDNA synthesis kit (Thermo Scientific, Waltham, MA, USA) using random hexamer. For qPCR, to the 1 × SYBR green (Roche, Mannheim, Germany) master mix added 1 µl of cDNA, 10 pM forward and reverse primers each were added and made up the final volume to 20 µl with nuclease-free water. The reaction for each primer was carried out in triplicates on ABI PRISM 7000 SDS with the following program: Initial denaturation at 95 ℃ for 3 min, followed by 40 cycles of 95 ℃ for 15 s and then annealing at 60 ℃ for 1 min. Dissociation curve analysis was performed from 60 ℃ to 95 ℃ to check the specificity of the reactions for each primer set. qRT-PCR analysis was performed with three biological replicates for each sample with GAPDH as the internal control. All the primers used in the study are listed in the Supplementary Table 1.
Table 1: Output of the transcriptome sequencing for the A. esculentus. View Table 1
Supplementary Table 1: List of primers and their amplicon size used in the study. View Supplementary Table 1
To obtain a high-quality leaf transcriptome of A. esculentus, four cDNA libraries were constructed and sequenced using Illumina Nextseq500. Sequencing yielded a total of 288.6 million raw reads. Adapter and low-quality reads were removed to obtain 206.3 million paired-end reads with a total of 28,871,205,535 nucleotides (nt) of 0.00% N and 45.2% GC content (Table 1). Further, high-quality trimmed reads were de novo assembled into 356,271 contigs (length ≥ 200) with the N50 of 1,657 nt, an average length of 1,015 nt, and a total of 361,767,712 nt. To minimize the redundancy in contigs, sequence clustering was performed with 70% similarity to produce two clusters and they were designated as unigenes. One cluster had unigenes over 70% similarity, and the other cluster was singletons. After clustering, reads with more than 400 nt were considered for further analysis. Thereby, we generated 66,382 unigenes with a total length of 71,350,824 nt and with an N50 of 1,408 nt and a mean length of 1,074 nt. Out of the 66,382 unigenes, there were 14,970 unigenes (22.6%) size ranging from 401-500 nt, 27,845 unigenes (41.91%) size ranging from 501-1000nt, and 23,567 unigenes (35.47%) with size more than 1000 nt (Table 2). Quality of the present de novo transcriptome assembly was evaluated by the length of assembled sequences with the previously reported transcriptome [10]. In the present dataset, about 35.47% of the assembled unigenes had length > 1000 nt, but it was only 0.79% with the already reported. This indicates that our assembled transcriptome could represent better sequence information (Table 2). Moreover, comparative analysis of both the transcriptome data showed that the number of longest unigenes, mean length, maximum length, and N50 sizes of our assembled unigenes were larger than the previously reported. Further, validation with 25 assembled transcripts between two transcriptome data indeed revealed the quality of the present transcriptome assembly (Supplementary Table 2). Furthermore, we have also found that some of the assembled transcripts length was longer than the reported length from the Malvaceae family members (Supplementary Table 3). These results depict that our sequencing data was better enough to assure the accuracy of the transcriptome assembly.
Table 2: Comparative statistics of assembled unigenes of present transcriptome dataset with the previously reported. View Table 2
Table 3: Statistics of SSR detected in A. esculentus. View Table 3
Supplementary Table 2: Comparative length analysis of the assembled transcripts between the present transcriptome data set and previously reported transcriptome. View Supplementary Table 2
Supplementary Table 3: Comparative length analysis of assembled unigenes from the present transcriptome dataset with the Malvaceae family members. View Supplementary Table 3
A. esculentus being a non-model plant without genome information, sequence-similarity based search was performed to annotate unigenes against UniProt database of green plants (Viridiplantae), with an E-value off ≤ 1.0e-05 and similarity of more than 30%. Among 66,382 unigenes, 37,121 (56%) of them showed homology with sequences in the UniProt database (Supplementary File 1). From the UniProt annotations, distribution of E-value, similarity, and species top hit were further analyzed, and the results were listed in Figure 1. The E-value distribution of the top hits in the UniProt database showed that 68.4% of mapped sequences had significant homology with the E-value < 1.0e-45, whereas 31.6% of sequences indicted moderate homology with E-value ranged from 1.0e-5 to 1.0e-45 (Figure 1a). The similarity distribution exhibited 45% of the query sequences with a similarity > 80%, while 55% of the hits had a similarity ranging from 30 to 80% (Figure 1b). Regarding the species distribution, we found majority of the annotated sequences were similar to Gossypium sps. (73.6%) followed by Theobroma cacao (13.2%), Corchorus capsularis (2.9%), and Corchorus olitorius (2.7%). All the four top BLAST hit in the analysis belonged to the Malvaceae family indicating the reliability of the assembly and annotation of unigenes (Figure 1c).
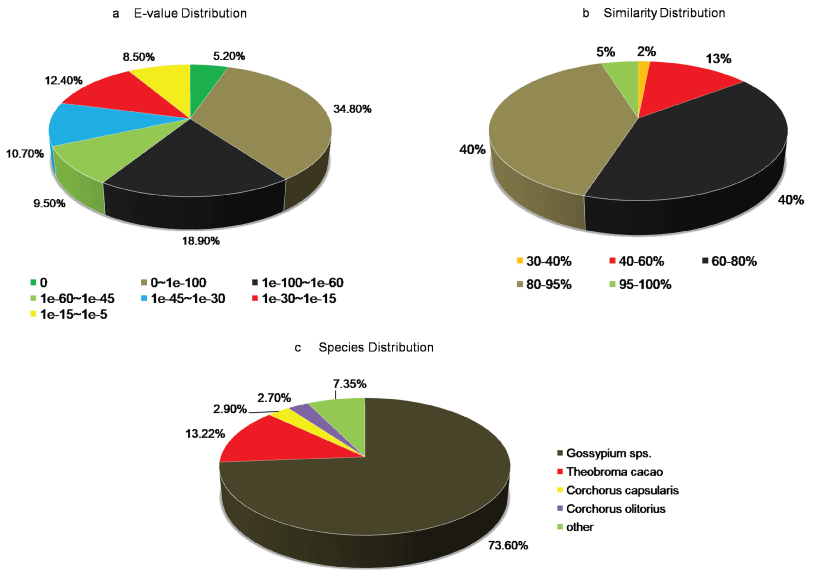 Figure 1: Characteristics of homology-based search of A. esculentus leaf transcriptome against the UniProt database. a) E-value distribution of BLAST hits for A. esculentus leaf transcriptome with E value cutoff ≤ 10-5; b) Sequence similarity distribution of top BLAST hits for A. esculentus assembled leaf transcriptome; c) Species distribution plot for the top hit sequences associated with unigenes of A. esculentus leaf transcriptome assembly.
View Figure 1
Figure 1: Characteristics of homology-based search of A. esculentus leaf transcriptome against the UniProt database. a) E-value distribution of BLAST hits for A. esculentus leaf transcriptome with E value cutoff ≤ 10-5; b) Sequence similarity distribution of top BLAST hits for A. esculentus assembled leaf transcriptome; c) Species distribution plot for the top hit sequences associated with unigenes of A. esculentus leaf transcriptome assembly.
View Figure 1
To facilitate the global analysis of A. esculentus leaf transcriptome, annotated unigenes were then assigned to GO terms for functional classification. About 76.9% (27,049) of the annotated unigenes were assigned to three main categories of GO classification, namely biological process, molecular function and cellular component (Supplementary File 1). Of them, assignments to the molecular function were the majority (21,353) followed by cellular component (14,124) and biological process (11,602). Under the category of molecular function, ATP binding (GO: 0005524, 3,757, 17.6%) and DNA binding (GO: 0003677, 2,832, 13.26%) were notably represented. Within biological process, the largest proportion was assigned to transcription DNA-templated (GO: 0006351, 1745, 15.04%) and metabolic process (GO: 0008152, 1,541, 13.28%). In the cellular component category, integral component of membrane (GO: 0016021, 7,941, 56.2%) and nucleus (GO: 0005634, 2,551, 18.06%) were the majority (Figure 2). To predict and categorize orthologous proteins, unigenes were aligned to the COG database. In total 8,233 unigenes (22.2%) were assigned to 24 functional categories of COG (Supplementary File 2). Among them, 'General function prediction only' (1,946 unigenes, 23.6%), and 'Translation, ribosome structure and biogenesis' (708 unigenes, 8.5%) related categories were notably represented (Figure 3).
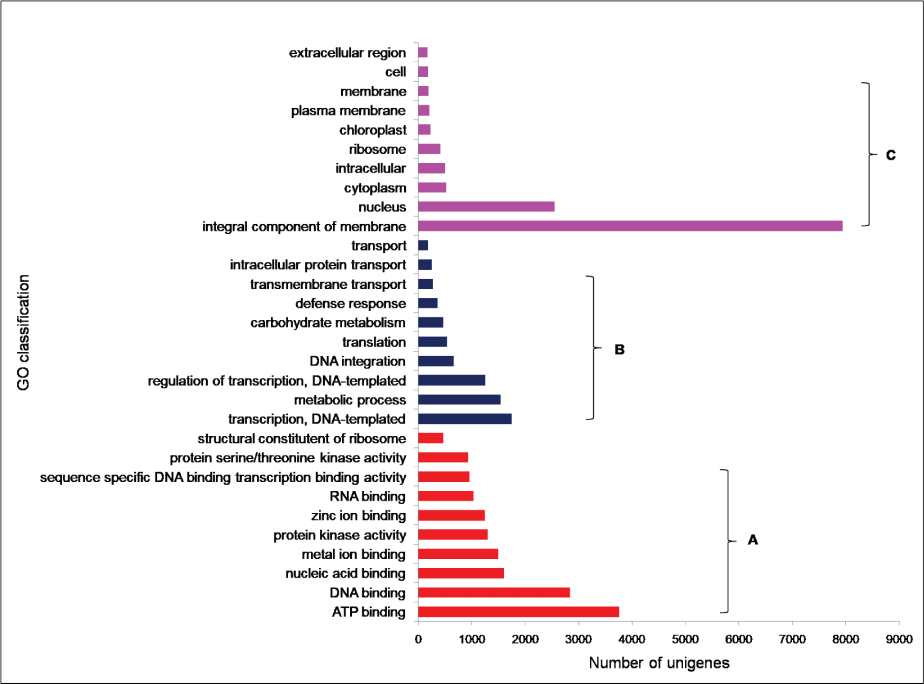 Figure 2: GO classifications of unigenes derived from A. esculentus leaf transcriptome. The results are shown as three main categories a) Molecular function; b) Biological function; c) Cellular component.
View Figure 2
Figure 2: GO classifications of unigenes derived from A. esculentus leaf transcriptome. The results are shown as three main categories a) Molecular function; b) Biological function; c) Cellular component.
View Figure 2
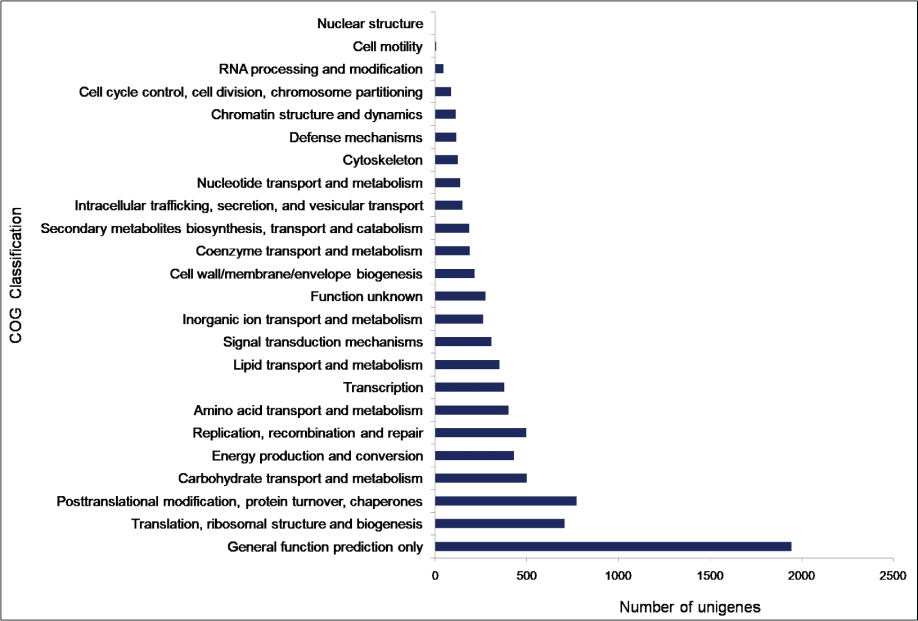 Figure 3: Clusters of Orthologous Groups (COG) classification of A. esculentus leaf transcriptome.
View Figure 3
Figure 3: Clusters of Orthologous Groups (COG) classification of A. esculentus leaf transcriptome.
View Figure 3
To identify the biological pathways active in A. esculentus, unigenes were mapped against Kyoto Encyclopedia of Genes and Genomes (KEGG) database. Out of 37,120 annotated unigenes, 16,307 (43.9%) had significant matches and were assigned to 143 KEGG pathways (Supplementary File 3). The five largest pathway groups were ribosome (ko03010, 735, 4.5%), plant-pathogen interaction (ko04626, 702, 4.3%), plant hormone signal transduction (ko04075, 647, 3.96%), carbon metabolism (ko01200, 559, 3.4%), and biosynthesis of amino acids (ko01230, 478, 2.9%) (Figure 4).
 Figure 4: Pathway assignment of A. esculentus leaf transcriptome based on KEGG. Unigenes were assigned into five categories of KEGG pathway a) Metabolism; b) Genetic information processing; c) Environmental information processing; d) Cellular processes; e) Organismal systems.
View Figure 4
Figure 4: Pathway assignment of A. esculentus leaf transcriptome based on KEGG. Unigenes were assigned into five categories of KEGG pathway a) Metabolism; b) Genetic information processing; c) Environmental information processing; d) Cellular processes; e) Organismal systems.
View Figure 4
From the KEGG pathway analysis of A. esculentus leaf transcriptome, KO numbers were retrieved and searched using KEGG mapper to visualize secondary metabolite pathway. Transcripts identified majorly in lignin (via. Phenylpropanoid pathway), flavonoids and terpenoids biosynthesis pathways were discussed.
Phenylpropanoids constitute a diverse group of plant-based secondary metabolites derived from phenylalanine [20]. Biosynthesis of phenylpropanoids starts with the formation of cinnamic acid from phenylalanine which leads to the formation of cinnamoyl-CoA, p-coumaroyl-CoA, caffeoyl-CoA, feruloyl-CoA, and sinapoyl-CoA. These CoA activated intermediates serves as the starting point for the synthesis of lignins, flavonoids, flavones, flavonols, anthocyanins, stilbenes, etc. In the present study, KEGG analyses of A. esculentus leaf transcriptome revealed 19 genes involved in the biosynthesis of different compounds of phenylpropanoid pathway (Figure 5).
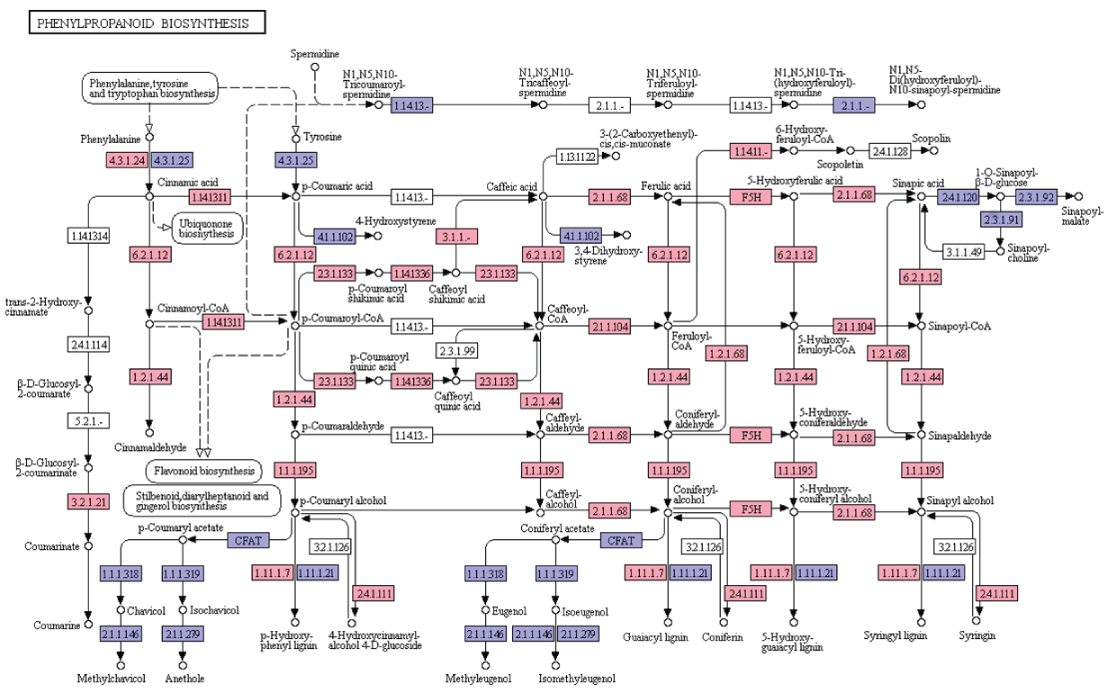 Figure 5: KEGG analysis of phenylpropanoid biosynthesis pathway genes in A. esculentus leaf transcriptome. Transcripts identified in the pathway are labeled in pink colour.
View Figure 5
Figure 5: KEGG analysis of phenylpropanoid biosynthesis pathway genes in A. esculentus leaf transcriptome. Transcripts identified in the pathway are labeled in pink colour.
View Figure 5
Lignin, one of the most important secondary metabolites with diverse functions in the plant growth, development and defense, is produced from phenylalanine/tyrosine metabolic pathway. Lignin monomers undergo a series of steps involving deamination, hydroxylation, methylation, and reduction in the cytoplasm and transported to the apoplast. Finally, lignin is polymerized with three types of monolignols (sinapyl alcohol, coniferyl alcohol, p-coumaryl alcohol) by peroxidase (POD) and laccase (LAC) [21]. In the present transcriptome dataset, most of the enzymes required for the synthesis of three monolignols were identified. They were phenylalanine ammonia lyase (EC: 4.3.1.24), trans-cinnamate 4-monooxygenase (EC: 1.14.13.11), 4-coumarate-CoA ligase (EC: 6.2.1.12), cinnamoyl-CoA reductase (EC: 1.2.1.44), cinnamyl-alcohol dehydrogenase (EC: 1.1.1.195), shikimate O-hydroxycinnamoyl transferase (EC: 2.3.1.133), coumaroylquinate 3'-monooxygenase (EC: 1.14.13.36), caffeoylshikimate esterase (EC: 3.1.1.-), caffeoyl-CoA O-methyltransferase (EC: 2.1.1.104), cinnamoyl-CoA reductase (EC: 1.2.1.44), cinnamyl-alcohol dehydrogenase (EC: 1.1.1.195), ferulate-5-hydroxylase (EC: 1.14.-.-), caffeic acid 3-0-methyltransferae (EC: 2.1.1.68). Later, synthesized sinapyl alcohol, coniferyl alcohol, and p-coumaryl alcohol are converted into syringyl lignin, guaiacyl lignin, and p-hydroxy-phenyl lignin by laccase and peroxidase (EC: 1.11.1.7) (Figure 5).
Flavonoids are diverse class of natural compounds, known as polyphenols that represent secondary metabolites from higher pants. They are classified into flavonols, flavones, flavanols, isoflavones, chalcones, catechins and their derivatives [22]. In the present study, we searched for the flavonoid biosynthesis genes and other related pathway genes from KEGG pathway analyses and are represented in the Figure 6. In the leaf transcriptome dataset of A. esculentus, starting from the initial enzymes of flavonoids biosynthesis (via phenylpropanoids pathway) like phenylalanine ammonia lyase (EC: 4.3.1.24), cinnamate 4-monooxygenase (EC: 1.14.13.11), 4-coumarate CoA ligase (EC: 6.2.1.12), and chalcone synthase (EC: 2.3.1.74) were observed. Besides, enzymes like shikimate O-hydroxycinnamoyl transferase (EC: 2.3.1.133), coumaroylquinate 3'-monooxygenase (EC: 1.14.13.36), and caffeoyl-CoA O-methyltransferase (EC: 2.1.1.104) required to convert p-coumaroyl-CoA to feruloyl-CoA were identified. Further, enzymes chalcone synthase (EC: 2.3.1.74), and chalcone isomerase (EC: 5.5.1.6) converts p-coumaroyl-CoA to naringenin were observed. Also, the enzymes required for naringenin conversion to produce eriodictyol and dihydrotricetin by flavonoid 3'5'-hydroxylase (EC: 1.14.14.81) and flavonoid 3'-monooxygenase (EC: 1.14.14.82) respectively were also detected. In addition to these, A. esculentus leaf transcriptome dataset also contained enzymes like bifunctional dihydroflavonol 4-reductase (EC: 1.1.1.219, EC: 1.1.1.234), naringenin 3-dioxygenase (EC: 1.14.11.9), flavonol synthase (EC: 1.14.20.6), anthocyanidin synthase (EC: 1.14.20.4), leucoanthocyanidin reductase (EC: 1.17.1.3), and anthocyanidin reductase (EC: 1.3.1.77) (Figure 6).
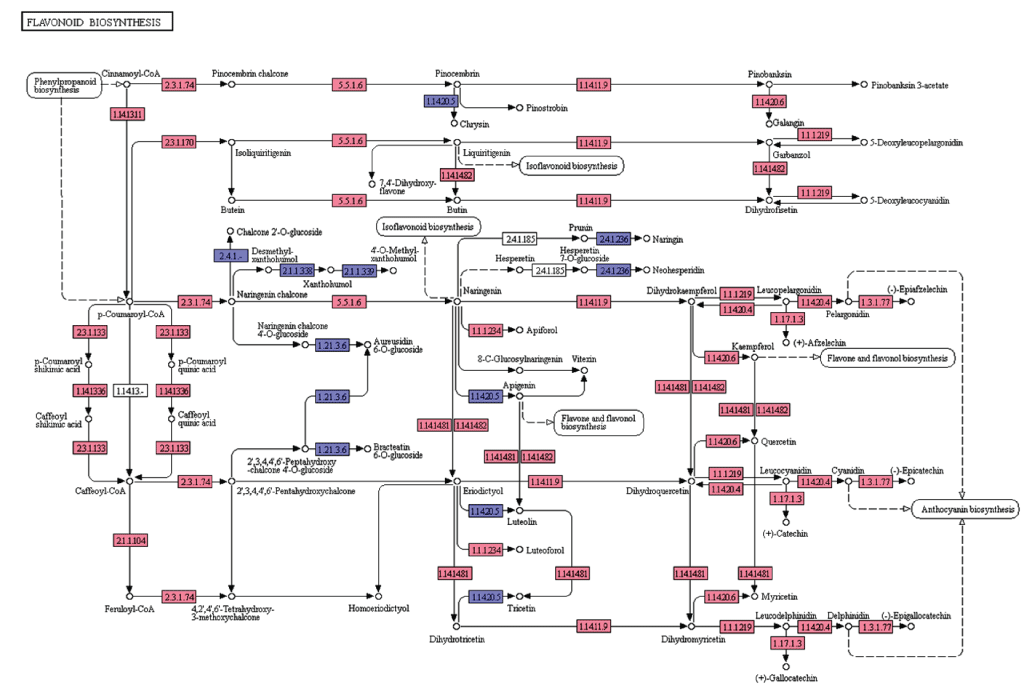 Figure 6: KEGG analysis of flavonoid biosynthesis pathway genes in A. esculentus leaf transcriptome. Transcripts identified in the pathway are labeled in pink colour.
View Figure 6
Figure 6: KEGG analysis of flavonoid biosynthesis pathway genes in A. esculentus leaf transcriptome. Transcripts identified in the pathway are labeled in pink colour.
View Figure 6
Terpenoids, major group of plant secondary metabolites, play an important role in plant-pathogen, plant-insect, plant-plant interactions [23]. Terpenoids are derived from geranyl pyrophosphate (GPP). GPP is produced through the cross talks between the mevalonate (MVA) pathway (cytosol) and 2-C-methyl-D-erythritol-4-phosphate (MEP) or DOXP pathway products (plastid) viz. isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP). Terpenoid biosynthesis is from both MVA and MEP pathways. MVA pathways begins with the formation of acetoacetyl-CoA whereas MEP pathway begins with D-glyceraldehyde 3-phosphate [24]. In the present study, 30 enzymes involved in the terpenoid biosynthesis were identified from the A. esculentus leaf transcriptome dataset. Transcripts identified in the MVA pathway were acetyl-CoA C-acetyltransferase (EC: 2.3.1.9), hydroxymethylglutaryl-CoA synthase (EC: 2.3.3.10), hydroxymethylglutaryl-CoA reductase (NADPH) (EC: 1.1.1.34), mevalonate kinase (EC: 2.7.1.36), phosphomevalonate kinase (EC: 2.7.4.2), diphosphomevalonate decarboxylase (EC: 4.1.1.33). MEP/DOXP pathway genes were 1-deoxy-D-xylulose-5-phosphate synthase (EC: 2.2.1.7), 1-deoxy-D-xylulose-5-phosphate reductoisomerase (EC: 1.1.1.267), 2-C-methyl-D-erythritol 4-phosphate cytidyl transferase (EC: 2.7.7.60), 4-diphosphocytidyl-2-C-methyl-D-erythritol kinase (EC: 2.7.1.148), 2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase (EC: 4.6.1.12), (E)-4-hydroxy-3-methylbut-2-enyl-diphosphate synthase (EC: 1.17.7.1, EC: 1.17.7.3), 4-hydroxy-3-methylbut-2-en-1-yl diphosphate reductase (EC: 1.17.7.4), isoprene synthase (EC: 4.2.3.27), farnesyl diphosphate synthase (EC: 2.5.1.1, EC: 2.5.1.10), and isopentenyl-diphosphate Delta-isomerase (EC:5.3.3.2). Other enzymes that were found in terpenoid backbone biosynthesis included geranylgeranyl diphosphate/geranylgeranyl-bacteriochlorophyllidea reductase (EC: 1.3.1.83, EC: 1.3.1.11), all-trans-nonaprenyl-diphosphate synthase (EC: 2.5.1.84, EC: 2.5.1.85), geranylgeranyl diphosphate synthase, type III (EC: 2.5.1.1, EC: 2.5.1.10, EC: 2.5.1.29), ditrans, polycis-polyprenyl diphosphate synthase (EC: 2.5.1.87), protein farnesyl transferase subunit beta (EC: 2.5.1.58), prenyl protein peptidase (EC: 3.4.22.-), STE24 endopeptidase (EC: 3.4.24.84), protein-S-isoprenylcysteine O-methyltransferase (EC: 2.1.1.100), prenylcysteine alpha-carboxyl methylesterase (EC: 3.1.1.-), prenylcysteine oxidase/farnesylcysteine lyase (EC: 1.8.3.5, EC: 1.8.3.6), NAD+-dependent farnesol dehydrogenase (EC: 1.1.1.354), and farnesol Kinase (EC: 2.7.1.216) (Figure 7). Transcripts identified in phenylpropanoids, flavonoids, and terpenoids biosynthesis pathways have been presented in Supplementary File 4. The sequence information about the secondary metabolite pathway genes identified in the present study will favour to genetically engineer this crop plant to improve is quality against various biotic and abiotic stress.
 Figure 7: KEGG analysis of terpenoid biosynthesis pathway genes in A. esculentus leaf transcriptome. Transcripts identified in the pathway are labeled in pink colour.
View Figure 7
Figure 7: KEGG analysis of terpenoid biosynthesis pathway genes in A. esculentus leaf transcriptome. Transcripts identified in the pathway are labeled in pink colour.
View Figure 7
Transcription factors play important roles in the regulation of various biological processes in plants. In this study, we identified a total of 4,041 transcription factor unigenes by comparing A. esculentus unigenes with the plant transcription factor database. The length of the transcription factor unigenes varied from 400-15,571 bp. The class > 1000 bp was the most enriched in total sequence number (68.74%), followed by 601-700 bp class (5.66%), 401-500 bp and 501-600 bp classes each by 5.34%, 701-800 bp class (5.07%), 901-1000 bp class (4.92%), and 801-900 bp class (4.89%) (Figure 8a). The potential transcription factors were distributed in 76 families, such as C2H2, WD40-like, CCHC(Zn), MYB-HB like, bHLH, AP2-EREBP, WRKY and so on (Figure 8b). Among these TF gene families C2H2, WD40-like, CCHC(Zn) were the most abundant transcription factor families (Supplementary File 5). Among the annotated transcription factor families, notable unigenes related to the secondary metabolism were bHLH, AP2-EREBP, WRKY, bZIP, C2C2-CO-like, MYB, Zf-HD, and ARF.
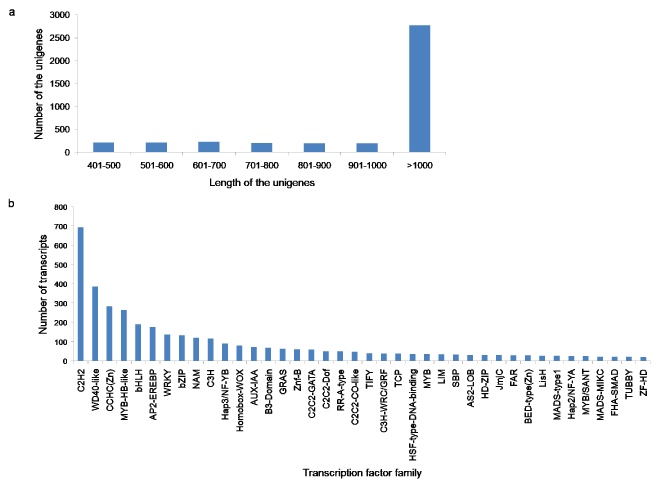 Figure 8: a,b) Identification of the transcripts encoding transcription factors in A. esculentus. (a) Length distribution of potential transcription factor genes (b) Transcripts distribution in various transcription factor families.
View Figure 8
Figure 8: a,b) Identification of the transcripts encoding transcription factors in A. esculentus. (a) Length distribution of potential transcription factor genes (b) Transcripts distribution in various transcription factor families.
View Figure 8
To identify potential SSRs, all the unigenes from the de novo assembly was searched using the MISA software. Our analysis detected a total of 9,578 SSRs in 66,382 unigenes, of which 933 unigenes had more than 1 SSRs (Table 3). Among repeat type classes, mono-nucleotide represented the largest fraction (57.78%) of all identified SSRs, followed by tri-nucleotides (21.28%), and di-nucleotides (19.2%). All the identified SSRs are shown in the Supplementary File 6. Although only a small fraction of tetra-nucleotides (147) and penta-nucleotides (18) were identified, the number is quite significant.
To experimentally assess the quality of de novo assembled unigenes, eight transcripts (Histone lysine methyltransferase, Nudix hydrolase, Polycomb group embryonic flower 2 like isoform, Retinoblastoma related protein, Adenosine kinase, DNA directed RNA polymerase IV and V subunit 4, Dicer-like protein, and Disease resistant protein) were randomly chosen for qRT-PCR analysis. DNase treated 1 µg of total RNA from the leaves of healthy bhendi plants was used in cDNA synthesis using random hexamer primers. Initally, PCR was performed with cDNA as the template for each transcript to confirm the specific amplification. Subsequently, qRT-PCR analysis was performed, and it showed the expression of all the selected eight transcripts with GAPDH as the internal control (Figure 9a and Figure 9b).
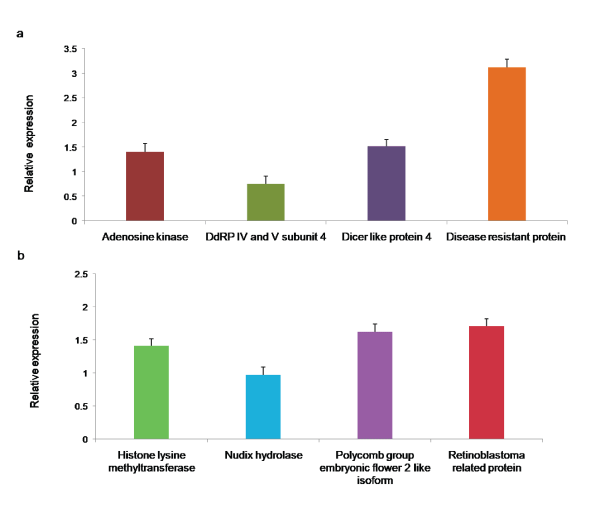 Figure 9: a,b): qRT PCR for the assembled A. esculentus leaf transcriptome. Primers were designed based on the assembled transcriptome for the eight selected transcripts. Their expressions were quantified by qRT-PCR with GAPDH as the reference. Each bar represents the mean ± SD of three biological replicates.
View Figure 9
Figure 9: a,b): qRT PCR for the assembled A. esculentus leaf transcriptome. Primers were designed based on the assembled transcriptome for the eight selected transcripts. Their expressions were quantified by qRT-PCR with GAPDH as the reference. Each bar represents the mean ± SD of three biological replicates.
View Figure 9
A. esculentus is an important annual vegetable crop. Its fleshy pod comprises the main edible portion of the plant with high nutrition and medicinal value. However, genome sequencing of this crop plant is not yet done probably due to the ploidy nature of chromosomes. Recent advances in the development of NGS technology, becomes the method of choice to obtain a large number of sequence information of non-model plants that lack genome information [25]. In 2013, Schafleitner, et al. [10] characterized A. esculentus transcriptome comprised of 153,414 unigenes with a maximum length of 20,230 bp and an N50 of 321 bp. N50 value is commonly used to assess an assembled transcriptome [26,27]. N50 value and the length of the reported A. esculentus transcriptome were lower when compared with other plants like chickpea, potato, turmeric, coconut, red raspberry, radish, and more due to the low quality and depth of sequencing [26,28-32]. Sequencing several independent libraries and merging their data for assembly is an ideal way to obtain high quality of assembled genome or transcriptome data with high coverage [33]. In accordance, de novo sequencing was performed from four independent cDNA libraries of healthy leaves and generated a total of 288 million paired-end raw reads. De novo assembly of clean reads using trinity produced a total of 356,271 contigs. Assembled sequences obtained from transcriptome assembly may be redundant due to alternative splicing events as well as misassemblies [34,35]. Therefore, duplicated sequences were removed from the contigs by CD-HIT clustering with 70% similarity. Further, selection of the longest transcripts in each cluster yielded 66,382 sequences with a maximum length of 38,471 bp and an N50 of 1,408 bp, which were considered as unigenes or transcripts. We observed a drop in the length of unigenes N50 with contigs. The most promising explanation is that most of the duplicated sequences are of longer length and were eventually dropped after CD-HIT analysis. Further, non-redundant unigenes were annotated using BLASTX against UniProt database of green plants. About 56% of the unigenes were annotated and there were also 29,262 (44%) unigenes that were not annotated. These unannotated sequences may represent transcripts that lack full-length domain or noncoding RNAs or misassemblies as reported with other plants [34,35,36]. Subsequently, functional annotation using GO, COG and KEGG showed the possible function of the assembled unigenes in various biological processes and biosynthetic pathway.
In the transcription factor analysis, 4,041 transcription factor unigenes were identified among 76 families with C2H2, WD40-like, and CCHC (Zn) as the most represented ones. C2H2 zinc finger proteins constitute a large gene family that participate in tissue and organ development in plants, especially root and floral development, as well as in biotic and abiotic response [37]. WD40-like proteins are shown to involve in various plant processes like floral development, light signalling, innate immunity and secondary metabolism [38,39]. CCHC zinc finger proteins bind with single-stranded DNA or RNA and initiates DNA replication [40]. Transcription factors bHLH, AP2-EREBP, WRKY, bZIP, C2C2-CO-like, MYB, Zf-HD, and ARF regulates various secondary metabolism pathway were identified from the A. esculentus leaf transcriptome. Similar results were identified in the leaf transcriptome of Phyllanthus amarus by Mazumdar and Chattopadhyay [41]. Recent study in maize shows that MYB transcription factor regulates the production of anthocyanins, and flavonoids in phenylpropanoids biosynthesis [42]. Transcription factors like bHLH, MYB, WD40, and WRKY are reported to regulate polyphenol biosynthesis in Tannat berries [43].
SSR markers serve as an important resource for studying functional genetic variation. We identified totally 9,578 potential SSRs from 66,382 unigenes. Previous studies on A. esculentus have identified more number of trinucleotide SSRs (492) with respect to other repeat type classes [10]. In our study, we could identify 2,039 trinucleotide SSRs. Identified SSRs in the present study could serve as potential genetic markers for the comparative genomics and population genetics studies across various species of Abelmoschus.
The transcriptome sequence information generated for A. esculentus in the present study will be useful for the investigation of differential gene expression and altered metabolic pathways during biotic and abiotic stresses for the improvement of this crop plant. For instance, wild rice plants (Oryza meyeriana) infected with Xanthomonas oryzae pv. oryzae showed resistance to the infection. Investigation of the RNA sequencing data indicated that the augmentation of defense response is due to the activation of various disease resistant genes by signal transduction pathways like phytohormones and ubiquitin-mediated proteolysis [44]. Lower expression of peroxidase super family genes contributed to the susceptibility of apple (Malus X domestica) plants against the Erwinia amylovora infection [45]. Response of apple transcriptome against various biotic stress conditions (fungal pathogens, virus, and bacteria) was studied by Balan, et al. [46]. They found that brassinosteroids genes were upregulated by funal pathogens, bacterial infection enhanced specific genes in sugar alcohol metabolism, gibberellins and jasmonates were strongly repressed by fungal and viral infections. Xu, et al. [47] identified the involvement of major transcripts related to the calcium signalling pathway during the salinity stress in pear plants. Similarly, transcript sequences related to the plant immune response or defense pathway can be selected from the present transcriptome data and their expressions can be studied in response to begomovirus infection which is considered as the major devasting pathogen contributing to the huge yield loss of A. esculentus [8,9,12,48]. Information acquired from the gene expression studies could be exploited to develop genetically resistant plants against various environmental cues.
In addition, sequence information from the transcriptome data can also be used to design exon capture arrays, to synthesize morpholino oligomers, to devise customized hybridization probes, to develop CRISPR/Cas9 cascade for the investigation of gene function in association with disease condition or with other qualitative trait for plants with no genome sequence available. Mukrimin, et al. [49] identified genetic variants of Norway spruce genes associated with susceptibility to Heterobasidion parviporum infection using exon capture approach. Sequence-specific morpholino oligomers are used to knock down gene mutation occurred during the diseased conditions [50]. Wang, et al. [51] showed enhanced resistance of rice against blast disease by CRISPR/Cas9-targeted mutagenesis of the ERF transcription factor. Recently, we have identified leaf specific 128 known and novel miRNAs from A. esculentus [12]. The sequence information from the present study could be utilized in the target identification and functional analysis of the reported novel miRNAs.
The comparative transcriptome analysis showed that the number and N50 sizes of the assembled unigenes from the present A. esculentus leaf transcriptome were larger than the previous reported transcriptome from Schafleitner, et al. However, very recently Zhang, et al. [52] reported A. esculentus transcriptome from root, stem, leaves, flowers, and fruits with 293,971 unigenes having N50 sizes of 1885 bp, which was better than our leaf transcriptome. The present, reported transcriptome assemblies along with precursors miRNA sequencing data [12] from A. esculentus can be merged to obtain comprehensive sequence information of this crop plant which will be useful for the functional genomics, host-pathogen interaction, and mRNA-miRNA network studies.
In conclusion, using Illumina sequencing technology, we generated de novo assembled reference A. esculentus leaf transcriptome, and its quality was assessed by the expression of the selected transcripts by qRT-PCR analysis. The transcriptome dataset presented in this study will serve as a valuable resource for molecular-genetics based research to develop increased resistance of this cultivar against various environmental cues. This study will provide an improved layout for the functional studies of genes and for the development of new markers for breeding purpose.
This work was supported by grants from SERB (Ref: No. SB/EMEQ-070/2013) and DBT (Ref: No. BT/PR2061/AGR/36/707/2011). The authors acknowledge Genotypic Pvt. Ltd. (Bangalore, India) for NGS sequencing facility. The authors are also thankful to Vempartham Suvekbala for proof reading the manuscript.