Extraintestinal manifestations of inflammatory bowel disease are common and extendable to all organs. Kidney and lower genitourinary system occurs in 4-23% of cases. This may be dependent on inflammatory bowel disease activity, secondary to metabolic disorders, drugs or others.
We present a case of a 68-year-old man with ulcerative colitis for 22 years admitted in our department for acute nephritic syndrome. Urinary microscopy suggested glomerular injury. A kidney biopsy was performed and was compatible with acute interstitial nephritis and IgA nephropathy. Toxicity of mesalazine and glomerulonephritis secondary to ulcerative colitis were assumed. The patient suspended mesalazine and started prednisolone with clinical improvement.
Our purpose is to sensitize the importance of having a prompt and thorough evaluation of acute kidney injury in patients with inflammatory bowel disease. We briefly review the broad spectrum of kidney manifestations in this population, focusing on mesalazine-induced nephrotoxicity.
Inflammatory bowel disease, Acute kidney injury, IgA nephropathy, Tubulointerstitial nephritis, Mesalazine
Inflammatory bowel disease (IBD) is a chronic inflammatory disease of the gastrointestinal tract, with its most frequent types being Crohn disease (CD) and ulcerative colitis (UC). Although its underlying mechanism is not entirely understood, abnormal T cell function has probably an important role [1].
Being a systemic inflammatory disease, extraintestinal manifestations are frequent, occurring in 6-47% of those patients. Kidney and lower genitourinary system are involved in 4-23% of patients. The most common renal manifestations include calculi, kidney tubular damage and parenchymal disease, like amyloidosis, tubulointerstitial nephritis, and glomerulonephritis [1-3].
The underlying mechanisms responsible for renal involvement in patients with IBD are not completely understood. However, it's believed to be related to several factors like chronic systemic inflammation, malnutrition and side effects of drugs. Most of those manifestations follow the clinical course of IBD, and their treatment is of great importance, as they may affect the patient's quality of life, morbidity, and mortality [3]. Apart from nephrotoxicity, the pathophysiological mechanisms of renal manifestations are classified in three major groups: Reactive manifestations (associated with gastrointestinal manifestations activity); manifestations independent of gastrointestinal disease activity (probably reflecting autoimmunity mechanisms); and metabolic manifestations (related to malabsorption or bacterial overgrowth) [2,3].
In 2014, Ambruzs, et al. published a large series of kidney biopsies from patients with IBD (83 patients). The most frequent indications for biopsy were acute or chronic kidney failure and nephrotic-range proteinuria. The most common histological diagnosis was IgA nephropathy (24%), followed by tubulointerstitial nephritis (TIN, 19%), arterionephrosclerosis (12%), acute tubular injury (8%), proliferative glomerulonephritis (GN, 7%), and minimal-change disease (5%). Most of the cases of interstitial nephritis had current or recent exposure to aminosalicylates [1].
One of the most well described renal manifestations of IBD is drug-induced nephrotoxicity in the form of interstitial nephritis, particularly to 5-aminosalicylic (ASA) and its derivatives like mesalamine or sulfasalazine. It's thought to be an idiosyncratic, delayed-type hypersensitivity, with no clear relationship between the duration or dose of those drugs and the development of interstitial nephritis [1,2]. Other medications associated with renal disease in patients with IBD include tumor necrosis factor-a (TNF-a) inhibitors and cyclosporine [3]. Although 5-ASA and its derivatives remain the main pharmacological culprits of TIN in IBD patients, some reports have shown an association of IBD activity and tubular proteinuria, and a correlation between tubulointerstitial damage and gastrointestinal disease activity [3].
Being a rare extraintestinal manifestation, a high index of suspicion is necessary to allow an early diagnosis of those glomerular and tubulointerstitial diseases. Therefore, monitoring kidney function is important in preventing, diagnosing, and reversing kidney damage, especially in patients on possible nephrotoxic medication [1].
Our purpose is to sensitize the importance of having a prompt, systematic and thorough evaluation of acute kidney injury in patients with inflammatory bowel disease. We briefly review the broad spectrum of kidney manifestations in this population, focusing on mesalazine-induced nephrotoxicity.
We present a 68-year-old white male with a medical history of UC for 22 years, partial thrombosis of the superior mesenteric and portal veins for 1 year - secondary to UC, under dabigatran; and benign prostatic hyperplasia, under finasteride.
Roughly eight weeks before his hospital admission, due to low UC activity, prednisolone was tapered off and the daily dose of mesalazine was doubled; at that time the patient had slightly decreased kidney function (serum creatinine (sCr) 0.95 mg/dL, glomerular filtration rate (GFR) of 81.9 mL/min/1.73 m2, using the CDK-EPI formula).
He was admitted for progressive anorexia, malaise and peripheral edema with one month of evolution. His vital signs were normal and besides bilateral peripheral edema, physical examination was unremarkable. Complementary tests revealed anemia (hemoglobin 10.7 g/dL) and acute kidney injury (AKI) (sCr 2.4 mg/dL) and the patient was transferred to the Nephrology Department for pursuing study. Additional study showed elevated erythrocyte sedimentation rate (102 mm/hour) and serum immunoglobulin (Ig) A (6,06 g/L; RV < 4.00); serum circulating immunocomplexes (89.31 RU/mL; RV < 20); mixed alkalosis and hypokalemia on arterial blood gas (pH 7.503, pCO2 26.2 mmHg, HCO3 22.9 mmHg, K 3.2 mEq/dL); urinary protein-to-creatinine ratio of 237 mg/g, urine specific gravity of 1,008 and leukoerythrocyturia on urinalysis (377/uL and 106/uL, respectively). Urinary sediment showed abundant dysmorphic erythrocytes (RBCs) and RBC casts under microscopy, with lipiduria and renal tubular epithelial cells (Figure 1). Further study, including monoclonal, polyclonal and infectious panel, was negative. Renal ultrasound excluded urinary calculi or obstruction. A kidney biopsy was performed. Under light microscopy, glomeruli revealed slight mesangial expansion and proliferation (Figure 2). The interstitial compartment showed mild mononuclear interstitial inflammatory infiltrate and fibrosis involving about 30% of the biopsy fragment (Figure 3). There was also acute tubular necrosis foci and tubular hematuria. No interstitial granulomas or crystal clefts were found. Congo red staining for amyloid was negative. Direct immunofluorescence examination of frozen renal tissue demonstrated mesangial deposits positive for IgA (++) and C3 (++) (Figure 4). These findings were consistent with interstitial nephritis due to salicylates and IgA nephropathy secondary to UC.
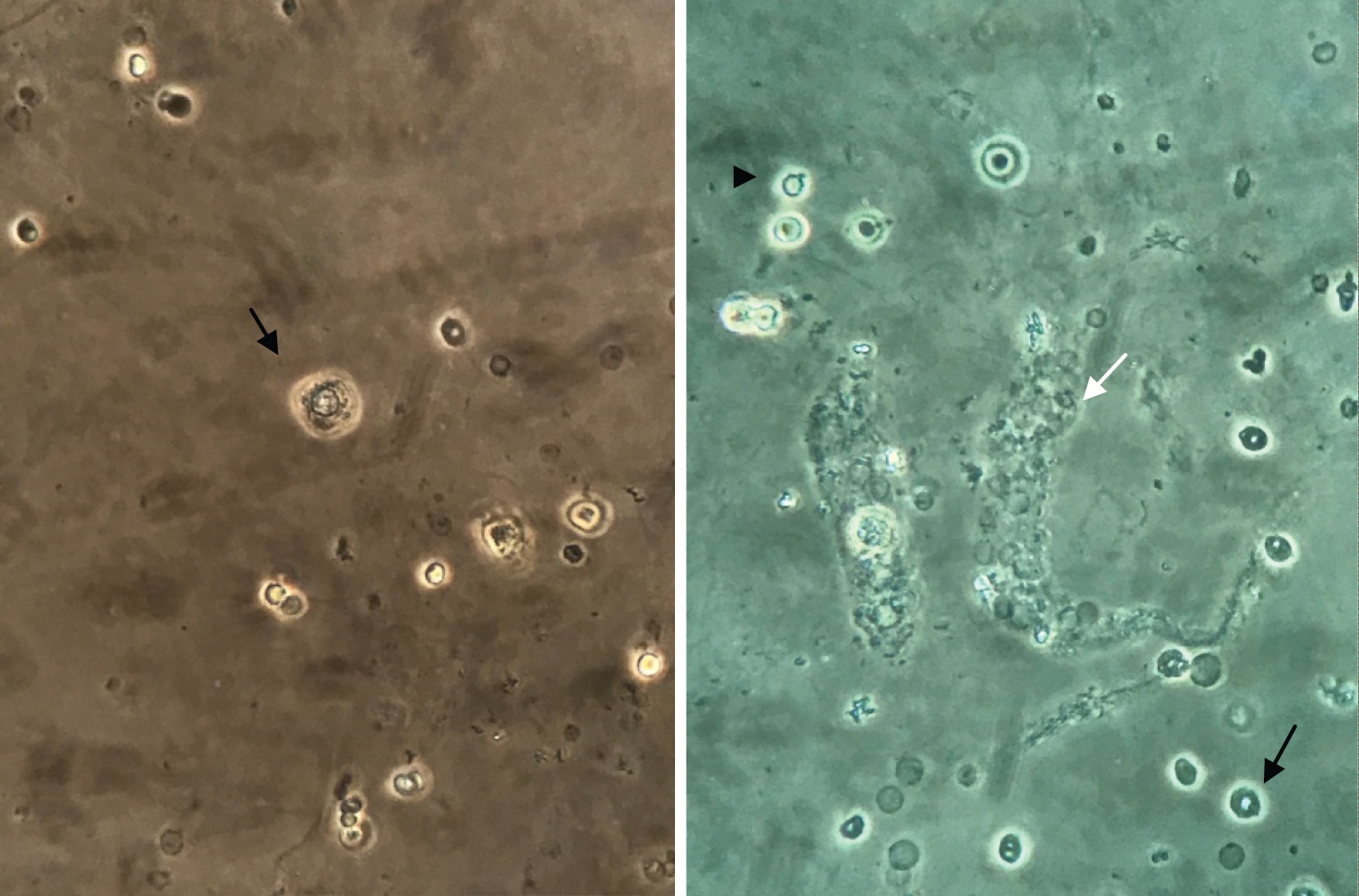 Figure 1: Urine microscopy (400x). (A) Renal tubular epithelial cell; (B) Erythrocyte cast (white arrow), dysmorphic glomerular erythrocytes (black arrow) and acanthocyte (arrow head).
View Figure 1
Figure 1: Urine microscopy (400x). (A) Renal tubular epithelial cell; (B) Erythrocyte cast (white arrow), dysmorphic glomerular erythrocytes (black arrow) and acanthocyte (arrow head).
View Figure 1
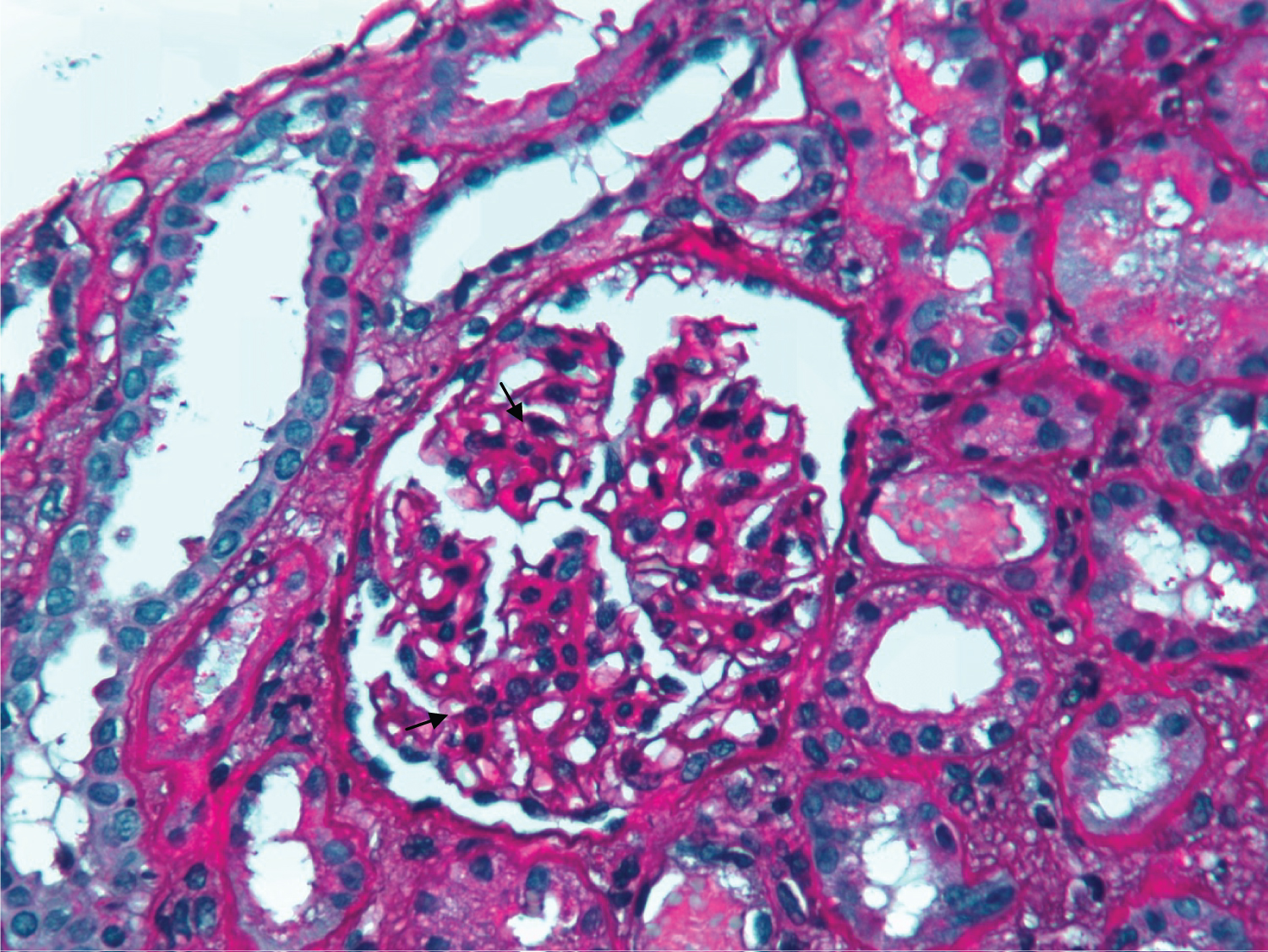 Figure 2: PAS x 400 - Slight mesangial expansion and proliferation on glomeruli (black arrows) and acute tubular necrosis foci (white arrow).
View Figure 2
Figure 2: PAS x 400 - Slight mesangial expansion and proliferation on glomeruli (black arrows) and acute tubular necrosis foci (white arrow).
View Figure 2
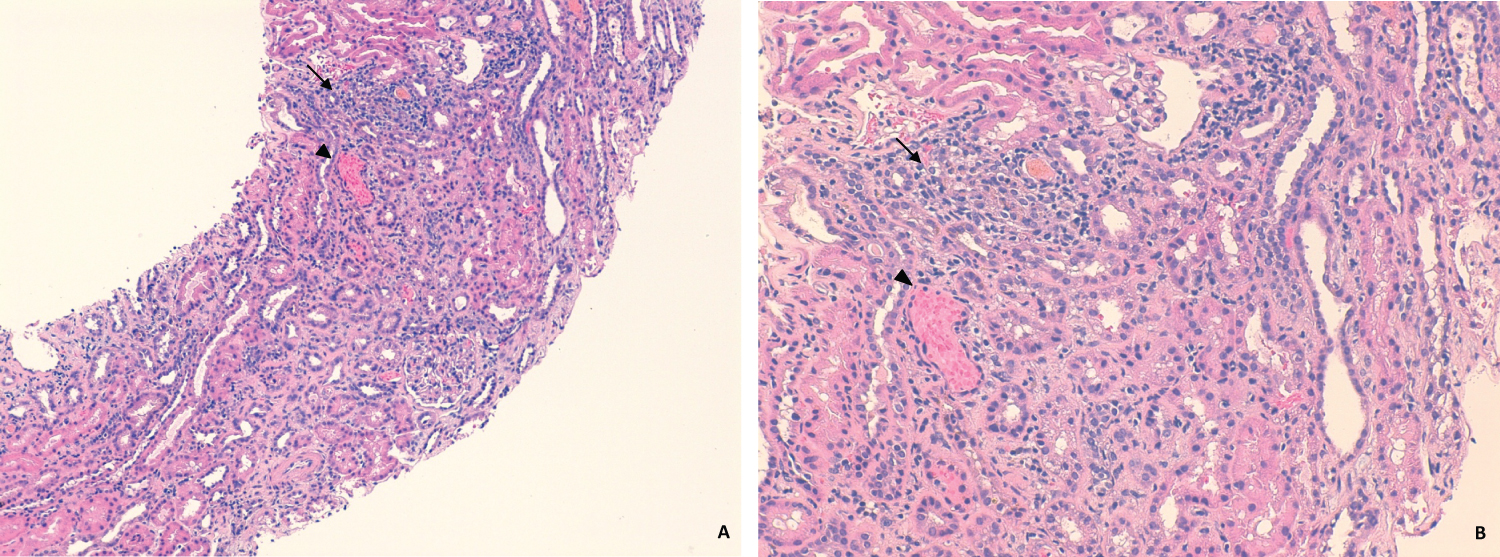 Figure 3: HE x120 (A) and x200 (B). Mild mononuclear interstitial inflammatory infiltrate on interstitial compartment (black arrow). Acute tubular necrosis foci (white arrow) and tubular hematuria (arrow-head) are also notorious.
View Figure 3
Figure 3: HE x120 (A) and x200 (B). Mild mononuclear interstitial inflammatory infiltrate on interstitial compartment (black arrow). Acute tubular necrosis foci (white arrow) and tubular hematuria (arrow-head) are also notorious.
View Figure 3
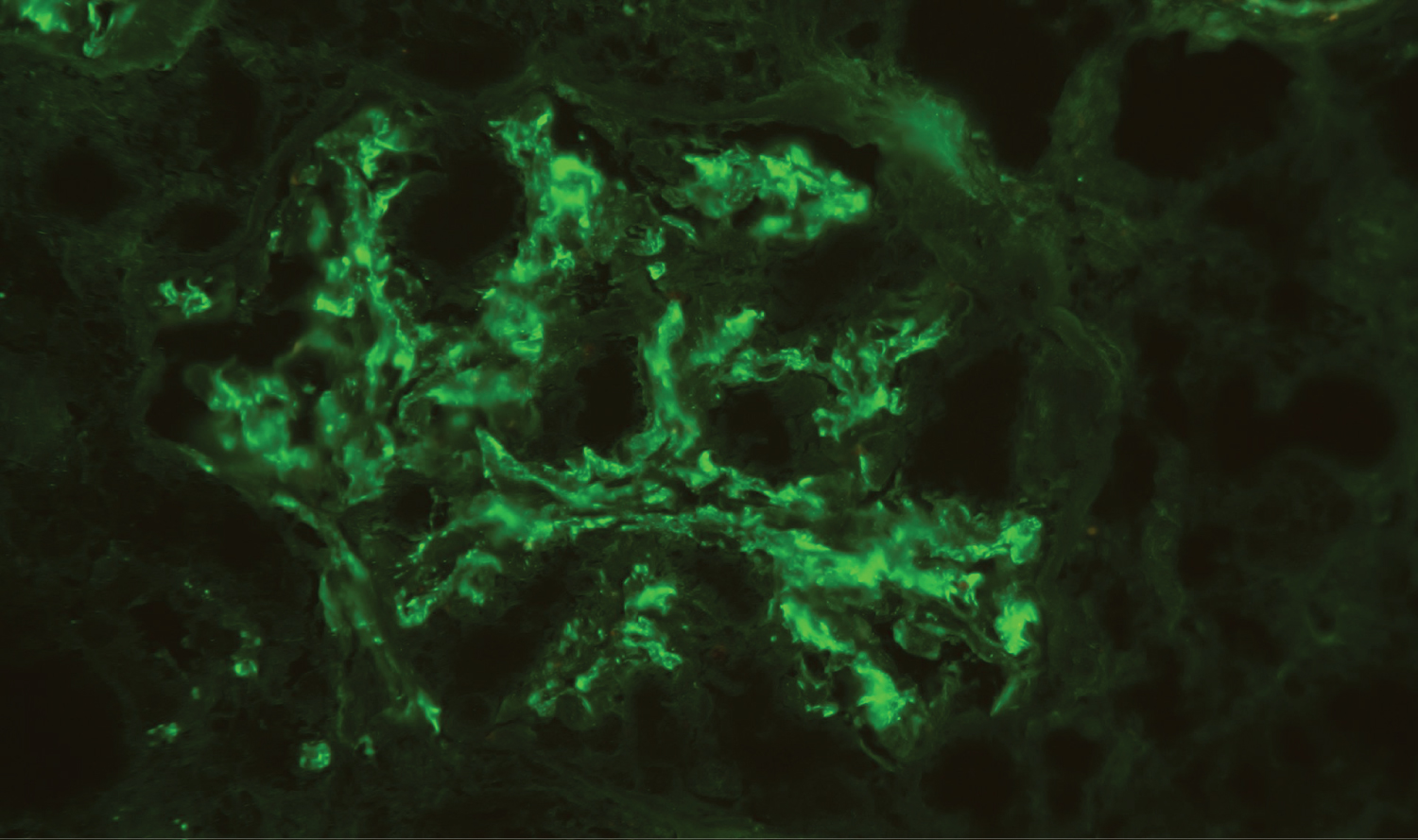 Figure 4: Direct immunofluorescence - Mesangial deposits of IgA (++). Deposits of C3 were also present (not showed). C1q, Kappa and Lamba were negative (not showed).
View Figure 4
Figure 4: Direct immunofluorescence - Mesangial deposits of IgA (++). Deposits of C3 were also present (not showed). C1q, Kappa and Lamba were negative (not showed).
View Figure 4
Mesalazine was discontinued and the patient was discharged on a prednisolone scheme of 1 mg/kg/day (60 mg) for two weeks with a gradual taper thereafter. Blood pressure control and low-salt diet were also optimized. During follow-up, our patient remained stable, with no recurrence of clinical symptoms. The patient's kidney function gradually improved to sCr 1.07 mg/dL, GFR 71 mL/min; urinary protein to creatinine ratio of 97.8 mg/g and erythrocyturia 23/uL and leukocyturia 6/uL at month 3, remaining stable after that.
Our patient presented with acute nephritic syndrome with AKI, low-grade proteinuria, leukocyturia and urinary sediment suggestive of active glomerular injury. Besides unspecific complaints that we previously reported, he had no other symptoms such as fever, arthralgia or skin rashes. He was normotensive, hydrated and denied macroscopic urinary findings. We promptly decided on kidney biopsy and the histopathological findings revealed out to be interesting. Acute interstitial nephritis predominated, which, given the history of mesalazine use, supported the diagnosis of drug-associated renal injury. Moreover, interestingly, other typical IBD-associated renal lesions, such as IgA nephropathy, ATN and tubular hematuria were also present.
Mesalazine is a 5-aminosalicylic acid (5-ASA) compound that has long been used as first-line therapy in IBD. As a lifetime maintenance treatment, considerations of long-term toxicity, in particular nephrotoxicity, which may be potentially irreversible, are utmost important [4]. Nonetheless the incidence of AKI associated with this agent has not been clearly determined.
Renal impairment may occur in up to 1 in each 100 patients treated but clinically significant AKI was estimated to occur only in 1 in 500 patients [2-5]. It may present as TIN or, rarely, as minimal-change nephropathy [3,6]. TIN clinical presentation is usually scanty and unspecific. Only a minority will have systemic symptoms like malaise, anorexia, weight loss, myalgia, fever, arthralgia or skin rash. Blood eosinophilia may be detected but also lacks specificity [2,5]. Most patients first present with asymptomatic increase in serum creatinine, which represents a late event [2,5]. Sterile leukocyturia and low-density urine gravity are common but non-specific features of immunoallergic TIN. Low-grade proteinuria is generally present but its value as early marker of 5-ASA-related-TIN is unreliable as it can be influenced by IBD activity [4,5,7]. Tubular enzymuria is a more sensitive and specific finding but it is not widely available [4,7]. Lastly, urinalysis can also reveal hematuria [5]. Recognizing that, and despite growing recognition of TIN associated erythrocyturia, it is our interpretation that abundant dysmorphic RBCs and RBC casts presented by our patient on urinary evaluation, are essentially associated to concomitant IgA nephropathy [8]. Gross microhematuria in its turn causes tubular precipitation and occlusion with subsequent ATN both also revealed in kidney biopsy and urinary sediment.
Potential 5-ASA- induced TIN may be diagnosed at any interval after beginning treatment. Fifty percent of patients present within the first 12 months, but it has also been observed after several years of treatment initiation [2,4,5]. The exact pathogenic mechanism of AKI is not completely known. The classic form of acute TIN, as we present, is thought to be a form of generalized delayed cell-mediated hypersensitivity reaction, characterized by mixed inflammatory cell infiltrate, composed of B and T lymphocytes and macrophages, and interstitial oedema [2,4-6]. In chronic stage, the extent of interstitial fibrosis and tubular atrophy will dictate irreversibility and progression to chronic kidney disease [5]. 5-ASA toxicity likely occurs as an idiosyncratic dose-independent phenomenon, unrelated to dosage or time of drug exposure [2,4-6]. Several authors suggest that undercurrent steroid therapy may mask AKI or delay presentation [4,5]. We believe that in our patient the reduction of prednisolone and the incremental dose of mesalazine both contributed to the development of significant renal damage.
Clinical suspicion or declared 5-ASA-induced-TIN should lead to prompt discontinuation of the drug. Recovery to baseline kidney function occurs in 85% of patients if the drug is stopped within 10 months of its initiation, but in only 33% if the time to diagnosis and drug withdrawal is longer than 18 months [2,4,6]. Albeit potentially reversible, it has been calculated that about 10% of patients with 5-ASA nephrotoxicity will progress to end-stage renal disease [2,4].
Steroids and azathioprine have also been used but the evidence for a beneficial role is not completely proven. A trial of high dose steroid (1 mg/day/kg, maximum 60 mg/day for up to 3 months) is recommended in patients whose renal function does not respond to drug withdrawal alone [2,4]. After discussion with Gastroenterology, given the severity of IBD presented by our patient and facing the risk of a new flare, we promptly decided on a dual strategy - of steroids and avoidance of the agent - instead of a stepped approach.
Regarding renal function monitoring in IBD patients treated with 5-ASA, the optimal schedule remains to be established. It has been suggested that sCr should be measured prior to commencing treatment, monthly for the first 3 months, 3-monthly for the remainder of the first year, and 6-monthly to annually thereafter. Patients receiving concurrent steroid therapy should be monitored more frequently [3,4,7]. To increase the likelihood of an early diagnosis, we authors also suggest periodic evaluation of routine automated urinalysis and, if a suspicion is present, the microscopic evaluation of the urine sediment. Moreover, all patient's new expositions, including drugs, and clinical intercurrences, namely IBD flares and infections, must be taken into account and should lead to an earlier assessment to exclude AKI.
Lastly, IgA nephropathy is the most encountered GN in patients in IBD patients by a wide margin [9]. An association between glomerular deposition of IgA and inflammation of the intestinal mucosa has been reported for many years suggesting a shared pathophysiology for these two entities [9,10]. However the responsible mechanisms have not been totally elucidated [10]. Most patients have increased numbers of IgA-secreting cells in the intestinal mucosa probably due to loss of mucosal integrity as a barrier to antigenic stimulation [10]. Therefore, secondary IgA nephropathy in IBD is likely to represent a complex axis of mucosal inflammation, loss of antigenic exclusion and tolerance, chronic immune stimulation and IgA overproduction [9]. The majority of patients show occurrence of IgA nephropathy during onset or exacerbation of IBD. Additionally, clinical remission of the kidney disease, sometimes with biopsy-proven clearance of mesangial IgA deposits, usually follows successful treatment of IBD [9,10].
The spectrum of kidney manifestations in UC patients can be broad. Parenchymal renal involvement is a rare but serious extra-intestinal manifestation. It can itself affect the glomerular, tubular or interstitial compartment. Coexistence, and documentation, of those three findings is rarely found. Prediction of specific renal affection, whether it is related to IBD, drug toxicity or others, sole based on clinical and laboratory findings is not linear. Kidney biopsy can clarify this diagnosis and thus guide the therapy and define the prognosis.
JCM and RDB were involved in the care and diagnosis of the patient, collected the data, performed the literature review and drafted the main manuscript. DBN interpreted the urinary microscopy. MVG and HSV interpreted the kidney biopsy. JLG, DBN, PMC, FR and FN were involved in the care and diagnosis of the patient, critically reviewed the manuscript, and interpreted the data.
The authors declare no conflict of interest.
Ethical issues including plagiarism, double publication, and redundancy have been completely observed by the authors.
None to be declared.