The mortality rate of Hemophagocytic Syndrome (HPS) is 26.5%-74.8%. Malignant neoplasms, hyperferritinemia, thrombocytopenia, older age, hypertriglyceridemia, and prolonged prothrombin are considered to be adverse prognostic factors. This study describes the underlying features of patient's survivors and non-survivors with HPS from one hospital between 2005-2019.
This is a retrospective study. We included patient with HPS diagnosis based on the HLH-2004 criteria, or who presented hemophagocytic cells in the bone marrow biopsy, or who had HPS diagnosis in the hospital discharge report. Demographic, clinical characteristics, underlying disorders and prognosis factors variables were collected. Kruskal Wallis, Fisher test and Mann-Whitney U test were used for the bivariate analysis. Kaplan-Meier method was utilized to calculate their survival rates.
Thirty patients were included. The median age was 55 years (± 18.3); 16 (53.3%) were female. The Hematological Malignancies (HM) subgroup had more and severe cytopenias [hemoglobin 6.5 (5.9-7.3; p 0.120), platelets 4500 (650-15,750; p 0.009), leukocytes 2050 (20-728; p 0.0001) and neutrophils 0 (0-280; p 0.002)]. The non-survivor group had a longer time of prolongation of the lNR [2.1 (1.2-3.7) versus 1.5 (1.1-1.6); p 0.028] and an older age at diagnosis of HPS [68 years (58.2-74.5) versus 40 years (34-57); p 0.043] versus the survivor group. The overall intrahospital mortality was 43.3%, being greater in HM subgroup [8 patients (66.7%); p 0.029].
The HM subgroup had a higher mortality, and a greater number and severe cytopenias. The non-survivor group had a longer time of INR and a higher age at the moment of diagnosis of HPS.
Hemophagocytic Syndrome, Prognostic Factors, Hemophagocytic Lymphohistiocytosis, Autoimmune diseases, Hematological Malignancies
HLH: Hemophagocytic Lymphohistiocytosis; HPS: Hemophagocytic Syndrome; HM: Hematological Malignancies; AI: Autoimmune; Inf.: Infections; MST: Malignant solid tumors; Ct: Chemotherapy; BM: Bone marrow; HC: Hemophagocytic cells; SLE: Systemic Lupus Erythematosus; ASD: Adult Still`s Disease; GM: Gliobastoma Multiforme; HIV: Human Immunodeficiency Virus; NK: Natural Killer; Hb: Hemoglobin; Pt: Platelets; Leu: Leukocytes; Neu: Neutrophils; Tg: Triglycerides; Fb: Fibrinogen; Fer.: Ferritin; Pre-hosp. stay dx: Hospital stay prior to diagnosis of HPS; AST: Aspartate aminotransferase; ALT: Alanine aminotransferase; LDH: Lactate dehydrogenase; T.B.: Total biluribin; INR: International normalized ratio; T.P: Total protein; A.P: Alkaline phosphatase; GGT: Gamma-glutamyl transferase; IL: Interleukin
Hemophagocytic Lymphohistiocytosis (HLH) is a clinical, inflammatory, acute and usually severe syndrome with high mortality without early treatment. The HLH is expressed by a proliferation and activation of T cells and macrophages, producing a hypercitokinemia [1,2]. HLH is also called Hemophagocytic Syndrome (HPS), a term and acronym that we will use to refer to this entity.
The HPS has been classified into 2 groups: Primary and secondary. The primary group, called Familial Hemophagocytic Lymphohistiocytosis, is a rare disease that commonly affects infants and children, and is inherited in an autosomal recessive manner [2,3,4]. The secondary group is associated with hematological malignancies (HM), autoimmune (AI) diseases, infections (Inf.), and other less common causes like secondary to bone marrow (BM) and solid organ transplants, malignant solid tumors (MST), pharmacological [immunosuppressors, biological treatments and chemotherapy (Ct)], and among others [1,5].
The HPS mortality rate is between 26.5% and 74.8% according to different published series [6-11]. In general malignant neoplasms underlying HPS, hyperferritinemia, thrombocytopenia, older age at diagnosis of HPS, hypertriglyceridemia, and prolonged prothrombin time greater than 3 seconds are considered to be adverse prognostic factors [7-16].
We are currently facing an entity with a high mortality without early treatment. The diagnosis and its therapeutic management are complex and are in the hands of different medical specialists, so understanding its various clinical manifestations, diagnostic criteria and adverse prognostic factors, are very important for its timely approach. In recent years, important advances have been published, such as the recommendations made by the American Society of Hematology and a scoring system (HScore) to predict the probability of diagnosing HPS made by the American College of Rheumatology, published in 2019 and 2014 respectively [5,17]. But, these advances leave the assessment of adverse prognostic factors, so the present study was an effort to determine this issue based on our own experience describing demographic, clinical manifestation, underlying disorders and laboratory findings to help us to evaluate the prognosis factors.
We retrospectively analyzed a cohort of 30 adult patients over 17-years-old with HPS diagnosis during their admission in the Donostia University Hospital between December 2005 and October 2019. The diagnosis of HPS was based on the HLH-2004 criteria, or if the patient presented HC in the BM biopsy, or who had HPS diagnosis in the hospital discharge report [4]. The diagnosis based on the HLH-2004 criteria required to meet at least five of the following criteria: 1- Prolonged fever (a temperature ≥ 38.5 ℃ for ≥ 7 days), 2- Splenomegaly (the costal margin exceeded 3 cm), 3 - Cytopenia involving in at least two lineages of peripheral blood (neutrophil count < 1.0 × 109/L, hemoglobin < 90 g/L or platelet < 100 × 109/L), 4- Hypertriglyceridemia (fasting triglycerides ≥ 3 mmol/L) and/or hypo-fibrinogenemia (fibrinogen ≤ 1.5 g/L), 5- Hemophagocytosis in bone marrow, spleen or lymph nodes, 6- Low or absent NK cells activity, 7- Serum ferritin ≥ 500 µg/L and 8- Soluble CD25 (soluble interleukin-2 receptor) ≥ 2400 IU/ml. However, the tests for soluble CD25 levels and NK cell activity were not available in our institution. In addition, HScore was calculated with the data collected from the medical record.
The study was approved by the ethics committee and given the retrospective informed consent was not obtained. However, after collecting the data, the patients were anonymized. In addition, the study followed the guidelines of the Organic Law 3/2018 of 5 December on the Protection of Personal Data and Guarantee of Digital Rights, which repealed Organic Law 15/1999 of 5 December on the Protection of Personal Data.
The demographic, clinical manifestation, etiologies, underlying disorders, diagnostic criteria, hospital stay measured in days [global hospital stay and hospital stay previous to the diagnosis of HPS (days from admission until the BM biopsy is performed and treatment was initiated)], prognostic factors, mortality, and relevant laboratory findings (complete blood count, biochemical profile, liver profile, coagulation, ferritin and others) were extracted from the electronic medical record.
Data analysis was performed using SPSS 21.0 software. Frequencies and percentages (n, %) were used to described the categorical variables. Continuous variables were described with the mean (x ± s) or median (RIQ) according to the degree of normality of the variable distribution. Non-parametric tests were used for most quantitative variables because the abnormal distribution. The analysis of inter-group differences (qualitative variables) was performed using the Fisher's test. Kruskal Wallis' test was used in the analysis between continuous quantitative variables and the polytomical categorical variable "etiology". The analysis between continuous quantitative variables and the analysis of the 2 groups (surviving and non-surviving) the Mann-Whitney U test were used. In addition, the Kaplan-Meier method was used to describe patient survival during hospital admission.
Thirty patients [14 man; mean age, 55.5 years (± 18.3 years)] with a diagnosis of HPS were included and divided into 5 subgroups (Table 1). The average HScore of the series was 270.5 (248.2-302.5), without finding statistically significant differences between the subgroups. In 25 patients (HM: 12, AI: 10, Inf.: 2 and MST: 1) An etiological diagnosis of HPS was found and in 5 cases (16.7%) no etiological diagnosis was found, despite multiple diagnostic tests such as blood tests, cultures and images. The debut of the primary etiology coincided with the appearance of HPS in 12 patients (AI: 6, HM: 4 and Inf.: 2). The coincidence of an infectious disease with HPS was observed in 10 patients, 2 were considered to be a primary etiology of HPS and 8 were probably the trigger of HPS [AI: 5 cases (2 Cytomegalovirus, 2 probably viral respiratory infections and 1 bacterial infection) and HO: 3 cases (2 Epstein Barr and 1 bacterial infection)].
Table 1: Etiologies and mortality of each subgroup of patients with Hemophagocytic Syndrome. View Table 1
All patients presented high fever, 53.3% splenomegaly and 33.3% hepatomegaly. Only one patient with SLE had neurological symptoms. The Table 2 and Table 3 shows the characteristics and comparative analysis of the demographic, clinical, laboratory findings, underlying disorders and mortality variables during hospital admission according to the etiological subgroup. Significant differences were observed in sex, age, leukocytes, neutrophils, platelets, transaminases and total proteins. Age at diagnosis of HPS was lower in the AI [40 (26.5-56.3)] and Inf. subgroups [45.5 (30-61); p 0.001)]. The HM subgroup had higher and more severe cytopenias [platelets 4500 (650-15,750; p 0.009), leukocytes 2050 (20-728; p 0.0001) and neutrophils 0 (0-280; p 0.002)] and low total proteins [4.3 (3.9- 4.5; p 0.003)]. The AI subgroup had the highest elevation of AST [457 (289-1140; p 0.026).
Table 2: Epidemiological characteristics, clinical manifestation, HScore, underlying disorders and mortality of each etiological subgroup of Hemophagocytic Syndrome during the admission. View Table 2
Table 3: Laboratory findings during hospital admission of each Hemophagocytic Syndrome subgroup. View Table 3
The survival of patients at 64 days during hospital admission was 50% (IC95%: 54.82-73.18) by the Kaplan-Meier method (Figure 1). The intrahospital mortality secondary to HPS was 43.3%, being statistically significant higher in the HM subgroup [8 patients (66.7%; p 0.029)]. Nine patients presented disseminated intravascular coagulation, of which 7 died (AI = 1/3, MH 6/6). Five patients were included in the HPS subgroup without defined etiology, 3 died during hospital admission and 2 continued to be followed by the Hematology service without finding the etiology.
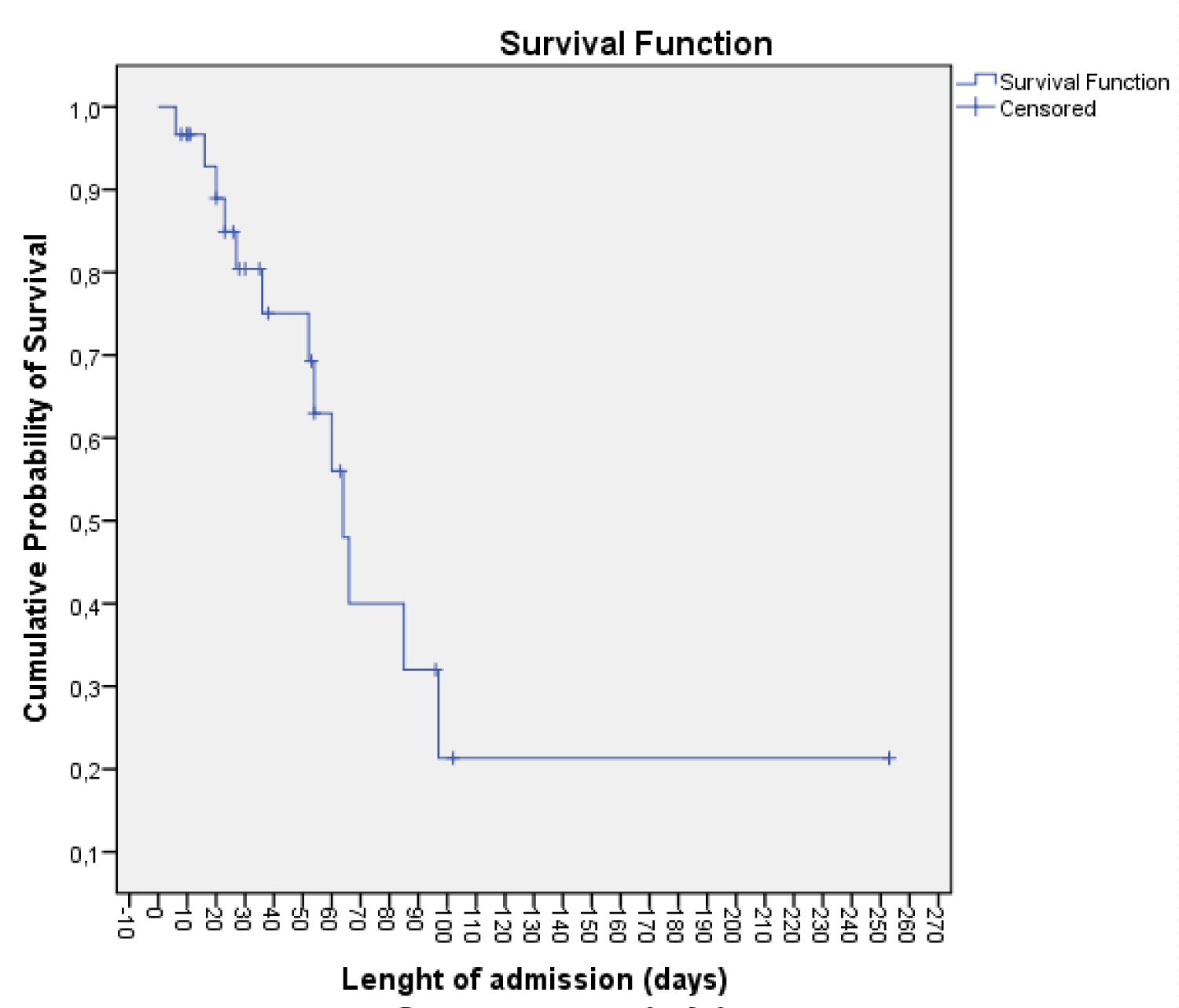 Figure 1: Analysis of survival during hospital admission by the Kaplan-Meier method.
View Figure 1
Figure 1: Analysis of survival during hospital admission by the Kaplan-Meier method.
View Figure 1
The non-survivors group of HPS presented a longer time of prolongation of the INR [2.1 (1.2-3.7) versus 1.5 (1.1- 1.6); p 0.028] and an age greater at the diagnosis of HPS [68 years (58.2-74.5) versus 40 years (34-57); p 0.043] compared to the survivors group (Table 4). There was no difference in the total platelet count, but all patients in the non-survivor group had thrombocytopenia [non-survivors: 13 (100%) vs. survivor 12 (70.6%); p 0.052)]. The survival group showed shorter length of hospital stay before treatment initiation than the non-survival group, but did not reach statistical significance. No significant differences were found in the number of comorbidities and others laboratory findings between the two groups. Seven patients (4 HM, 2 AI and 1 HPS without defined etiology) were admitted to the Intensive Care Unit (ICU), of which 4 died (3 HM and 1 HPS without defined etiology) (p = 0.666).
Table 4: Demographic characteristics, clinical manifestations, laboratory findings and underlying disorders between the haemophagocytic syndrome survivor and non-survivor group. View Table 4
After hospital discharge 4 of 16 patients that we follow up died due to their primary etiology: Non-Hodgkin's lymphoma (6.8 months after discharge), myelodysplastic syndrome (43.9 months after discharge), multiple myeloma (12 months after discharge) and GM (1 month after discharge).
In our study the most frequent causes of HPS were HM and AI diseases, similar to others published series. [3,7,12,18]. The most frequent cause of HPS found were lymphoma, SLE and ASD, as described in most of the series AI or HM diseases [8]. HPS can also occur in the course of other hematological malignancies, such as B-cell lymphoma, Hodgkin's lymphoma, acute and chronic leukemias, acute myeloid leukemia, and myelodysplastic syndromes, as in our study [10,17]. In some series the absence of an etiological diagnosis of HPS was 30% [10]. In our series, the absence of an etiology was 16.7%, with a mortality rate of 60% (p 0.029). The absence of an etiological diagnosis of HPS probably delayed the initiation of treatment and influenced in the prognosis. In our series we found only one case of HPS secondary to temozolomide, a chemotherapeutic agent. The diagnosis of HPS secondary to CT is complicated, probably by pre-existing neutropenia; in our opinion, the diagnosis of HPS should be considered in patients which continues with neutropenia and fever after stopping the CT and do not respond to broad-spectrum antibiotics.
The clinical features reflect the alteration of the immune system, induced by a hypercitokinaemia with increased interferon-gamma, tumor necrosis factor alpha, interleukins (IL) 1, IL-6, IL-10, IL-12, IL-18 and macrophage colony-stimulating factor, released by highly activated lymphocytes and macrophages [19,20]. The clinical and analytical features that suggest HPS are prolonged fever without response to antibiotherapy, hepatosplenomegaly, thrombocytopenia progressing to severe pancytopenia, hyperferritinemia, hypertransaminasemia, hypofibrinogenemia, hypertriglyceridemia, and hypoalbuminemia [21]. In our series we observed that the HM subgroup presented a greater number and severity of cytopenias. The triglycerides may not increase until the liver has been damaged for a long period of time; this means that hypertriglyceridemia is a sign of severe and late liver failure. In our study, there were no differences between the etiological subgroups or by survival group, unlike other studies, where hypertriglyceridemia proved to be a predictor of adverse outcome [14]. This may be because most of our patients in the survival group were patients with AI disease, who had lower mortality and greater liver alteration.
As we have already mentioned there is a great variability in the mortality rate according to the studies published. In our study the overall mortality was 43.3%, being higher in the subgroup HM (66.7%; p 0.029). We observed in the group of non-survivors, a greater age at the time of diagnosis of HPS and a greater prolongation of the INR. In others studies the mortality rate of HPS secondary to AI diseases was lower, followed by infectious diseases and primary HLH. In general, the malignancies underlying HPS, especially lymphoma, are considered an adverse prognostic factor [7,11,12,14].
The thrombocytopenia is considered an adverse prognostic factor in HPS, and it's probably the most important one of all [9,11,22,23]. In our study the HM subgroup had lower levels of platelets [4,500 (650-15,750; p 0.004)]. In the analysis between the non-survivor and survivor groups there was no statistically significant difference, but all patients of the non-survivor group had thrombocytopenia in contrast to the survivor group. There are several mechanisms likely to cause thrombocytopenia: decreased production by insufficient BM, overconsumption caused by disseminated intravascular coagulopathy and hypersplenism. Currently, there is substantial evidence supporting the association between platelets and inflammation. Platelets express Toll-like receptors, indicating an ability to directly attract leukocyte-like microbial pathogens [24]. They bind to von Willebrand factor attached to endothelial cells, causing leukocytes to accumulate on the endothelial surface [25]. In addition, platelets have a reciprocal relationship with the complement system, which helps eliminate infection [26,27]. In summary, thrombocytopenia results in an increased risk of bleeding, impaired immune response, and bone marrow failure, suggesting a worse prognosis.
Hyperferritinemia was reported an independent prognostic variable of mortality in HPS [15,28]. The ferritin has been previously reported that its level greater than 10,000 µg/L have 90% sensitive and 96% specific in pediatric HLH. [29]. Almost all adults with HPS meet the diagnostic criterion of serum ferritin greater than 500 µg/L, the sensitivity and specificity of this marker in the adult population is much less impressive [30]. A review showed that a hyperferritinemia over 50,000 mg/L was not seen most often in adult HLH patients, but in patients with renal failure, hepatocellular injury, infections and hematological malignancies [31]. In our study we do not found significant difference in the comparative analysis of the etiologies subgroup and survivor groups.
The age is also considered an independent adverse prognostic factor for HPS. In our study the non-survivor group had an older age, which is consistent with other studies [14-16]. This may be related to senescent organs resulting from more severe organ dysfunction [22]. It should be mentioned that the non-survivor group in our study did not present a greater number of comorbidities than the survivor group, so it appears that presenting comorbidities does not influence in the prognosis.
In relation to coagulation, it has been observed in other studies that prolongation of the prothrombin time greater than 3 seconds is associated with a worse prognosis. In our study a prolonged INR was observed in the non-survivor group (p 0.028) [13,22]. The INR provides a qualitative assessment of anticoagulation activity, and the prolongation is a risk factor for bleeding events.
The mortality of HPS from all causes used to be approximately 95% with a median survival of 1-2 months, before the initiation of targeted therapy. In general, current therapy is based in the combination of immunosuppressant's and cytotoxics to stop the hyperinflammatory state, whatever the cause, but the main goal of therapy is to treat the underlying disease. The induction treatment in the protocols HLH-94 and HLH-2004 are dexamethasone and etoposide, and for maintenance cyclosporine A. In refractory or recurrent cases, intrathecal methotrexate and allogeneic hematopoietic cell transplantation are used [4,32,33]. In the specific case of Macrophage Syndrome (HPS secondary to AI disease) the main treatment are the glucocorticoids. In refractory cases to glucocorticoids, cyclosporine A 2-7 mg/kg/day is recommended to add [34-36]. In refractory cases of these two treatments, the HLH-2004 protocol should be considered. However, etoposide is not recommended because the serious side effects [37]. Biological therapies, such as IL-1 and IL-6 inhibitors, is recommended in refractory cases [38,39]. Others monoclonal antibodies and Janus kinase pathway inhibitors show promise of being effective. Emapalumab is a human monoclonal antibody directed against interferon-gamma, and is the first Food and Drug Administration approved therapy for primary HLH, but is under regulatory review in the European Union. Emapalumab is approved for treatment of primary HLH that is refractory, recurrent, progressing or intolerant to current HLH treatments in both adult and pediatric patients [40]. Ruxolitinib, a Janus kinase 1/2 inhibitor, can be option as a front-line therapy in children with secondary HLH. Also, treatment with low dose ruxolitinib plus HLH-94 protocol might be a potential choice for secondary HLH [41,42]. Other therapies such as intravenous immunoglobulin, cyclophosphamide, and plasmapheresis have provided inconsistent results.
The HM subgroup had greater mortality and more number and severe cytopenias than the other subgroups. The non-survivor group presented a longer time of INR and a higher age at the time of diagnosis HPS compared to the survivor group. The subgroup of HPS without defined etiology presented high mortality, maybe due to a delay in diagnosis and therefore in treatment.
The study was approved by the ethics committee of the Donostia University Hospital, and given the retrospective nature of this study; informed consent was not obtained. However, after data collection, patients were anonymized. In addition, the study followed the guidelines of the Organic Law 3/2018, of December 5, on the Protection of Personal Data and Guarantee of Digital Rights, which repealed the Organic Law 15/1999, of December 5, on the Protection of Personal Data in Spain.
Consent was not obtained because the retrospective nature of the study.
Not applicable.
The authors declare that they have no competing interests.
Funding research wasn't received.
CAED: Main author. Major contributor in writing the manuscript.
JCA: Second main author. Contributor in writing and review the manuscript.
PCM: Third main author. Performed the statistical analysis and review the manuscript.
ADDS: Helped to make the introduction and to collected information from medical records.
JRFS: Performed the reading and analysis of the bone marrows and collected information from medical records.
NAL, JAVJ, LMLD, JJCF, OMA and EUI: Collected information from medical records
JMB: Reviewed the manuscript.
Also, all authors read and approved the final manuscript.
Acknowledgements: Not applicable.