Paraneoplastic Pemphigus (PNP) is an autoimmune-related acquired bullous disease associated with neoplasia. Both humoral and cellular immunity are involved in the pathogenesis of PNP. Characteristically, PNP has a diverse spectrum of clinical and immunopathological features. Suprabasal acantholysis and clefts with scattered necrotic keratinocytes are the unique histopathological features. PNP patient sera recognize multiple antigens, which have been identified as the plakin protein family. Non-Hodgkin's lymphoma, Castleman's disease, thymoma, follicular dendritic cell sarcoma and chronic lymphocytic leukemia are the commonly associated neoplasms in PNP. The treatment of PNP to date has been rather disappointing. For patients with Castleman's disease, the early detection and removal of the tumor are critical for the treatment of PNP.
Paraneoplastic pemphigus, Stomatitis, Direct immunofluorescence, Indirect immunofluorescence, Rat urinary bladder, Plakin protein, Non-Hodgkin's lymphoma, Castleman's tumor, Thymoma, Follicular dendritic cell sarcoma, Chronic lymphocytic leukemia, Corticosteroids, Resection
Paraneoplastic Pemphigus (PNP) is a debilitating and lethal adult and pediatric autoimmune blistering skin disease that occurs in the setting of a known or occult neoplasm especially lymphoproliferative neoplasms [1]. In 1990, Anhalt, et al. [2] described five atypical pemphigus cases which were associated with lymphoproliferative disease. Anhalt called this disease paraneoplastic pemphigus. The term paraneoplastic autoimmune multiorgan syndrome (PAMS) was suggested later by Nguyen, et al., given that is not a skin disease, but a syndrome characterized by the presence of mucocutaneous and non-cutaneous pathology associated with neoplasia [3]. More than 260 case reports of PNP have been documented since that time [1,4,5] and about 20 of the reported patients were children or adolescents. Yet PNP may be more common than is generally believed given that serum detection of autoantibodies is only performed by a handful of laboratories. One clinical sign is severe stomatitis, although other skin changes can vary considerably. Suprabasal acantholysis and clefts with scattered necrotic keratinocytes are the unique histopathological features. Autoantibody activity in PNP is complex, but usually highly characteristic [2,6-8]. PNP patient sera recognize multiple antigens, which have been identified as the plakin protein family that includes desmoplakin, bullous pemphigoid antigen I, envoplakin and periplakin, and desmogleins 1 and 3. Non-Hodgkin's lymphoma, Castleman's tumor, thymoma, follicular dendritic cell sarcoma and chronic lymphocytic leukemia are the commonly associated neoplasms in PNP [2]. These clinical characteristics, in addition to positive immunophenotypic findings make the diagnosis of PNP. The treatment of PNP to date has been rather disappointing. The mortality rate associated with the disease is high and common causes of death include sepsis, gastrointestinal bleeding, and bronchiolitis obliterans [6,9]. The aim of this article was to evaluate the clinical, pathological and immunological characteristics of PNP based on the analysis of literature reports.
The clinical presentation of PNP, although highly varied, typically begins with a florid, painful eruption of mucous membrane lesions, which may involve the oropharynx, nasopharynx, tongue, vermillion of the lips (Figure 1), conjunctiva, anogenital region (Figure 2), or esophagus [1,4,10,11]. Oral mucosal lesions were usually more extensive than that of pemphigus vulgaris or erythema multiforme. In addition to conjunctivitis, ocular presentation also included corneal ulceration, which was successfully managed with topical tacrolimus, cyclosporine and systemic methylprednisone, thalidomide, methotrexate in a case reported [12]. Severe stomatitis can be one of the hallmarks of PNP.
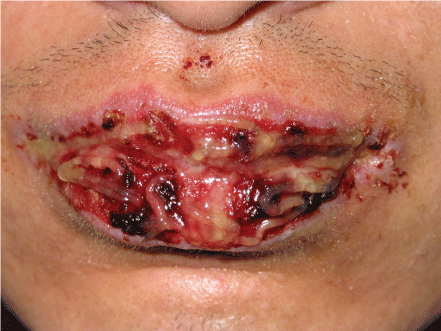 Figure 1: Severe erosion, ulcer and crusts on lips. View Figure 1
Figure 1: Severe erosion, ulcer and crusts on lips. View Figure 1
 Figure 2: Severe erosion, ulcer on prepuce. View Figure 2
Figure 2: Severe erosion, ulcer on prepuce. View Figure 2
The onset of cutaneous lesions has a variable delay after the appearance of mucous membrane lesions, ranging from days to months [1,2,4]. Cutaneous features of PNP are polymorphic, including vesicles, blisters, erosions, patches, papules and plaques. The Nikolsky sign may be absent. The heterogeneity in the presentation of the cutaneous manifestations of this disease has led to a stratification of PNP based on variable and polymorphic features that mimicked Pemphigus Vulgaris (PV), Pemphigus Foliaceus (PF), Bullous Pemphigoid (BP), Erythema Multiforme (EM), lichen planus and Graft-versus-Host Disease-like [11]. Specifically, (i) Pemphigus-like: superficial vesicules, flaccid blisters, erosions and crusts, occasional and limited erythema; (ii) Bullous pemphigoid like: scaling erythematous papules that may be associated or not with tense blisters; (iii) Erythema multiforme-like: polymorphic lesions, mainly scaling erythematous papules with erosions or occasionally ulcers with difficult healing; (iv) Lichen planus-like: small squamous flat-topped violaceus papules and intense involvement of mucosal membranes; (v) Graft-versus-Host Disease-like: disseminated dusky red scaly papules [3]. These cutaneous manifestations often present in waves of lesions and, in contrast to pemphigus vulgaris, blisters in PNP do not arise from normal skin but typically develop from inflammatory papules or macules, which greatly outnumber bullous eruptions in PNP on individual patients [13]. Cutaneous syndromes are characterized by blister formation due to an autoimmune response to components of the epidermis or basement membrane in the context of a neoplasm.
There has also been well-documented evidence of involvement of internal organs, such as lungs, smooth muscle, thyroid, kidney, and gastrointestinal tract [11]. Bronchiolitis Obliterans (BO) is the most common complication and progressive respiratory failure caused by BO is the most important cause of death. Evidence to date indicates that in paraneoplastic pemphigus, autoantibodies directed against plakin proteins may be responsible for acantholytic changes in the respiratory epithelium, among which, epiplakin and desmoglein3 might be the major autoantigens [14-16]. Moreover, there is an increased occurrence of Castleman's disease in patients with pulmonary complications [17]. Myasthenia Gravis (MG) is a complication of PNP related to its neurological involvement. A recent research [18] about MG and its correlation in patients with PNP revealed that overall 39% of patients with PNP experienced muscle weakness, 35% were diagnosed with MG, and MG complications did not affect the overall survival percentage in PNP. Another term for PNP, Paraneoplastic Autoimmune Multiorgan Syndrome (PAMS) has been proposed by Nguyen, et al., given that it is not a skin disease, but a syndrome characterized by the presence of mucocutaneous and non-cutaneous pathology associated with neoplasia [3]; and in PNP the autoantibodies bind in the kidney, as well as to the epithelium of colon, the small intestine, thyroid, striated muscle and smooth muscle [11].
The histologic findings from patients with different clinical presentations of PNP have shown similarities with other known dermatologic conditions (Figure 3), perhaps indicating commonalities in pathogenesis.
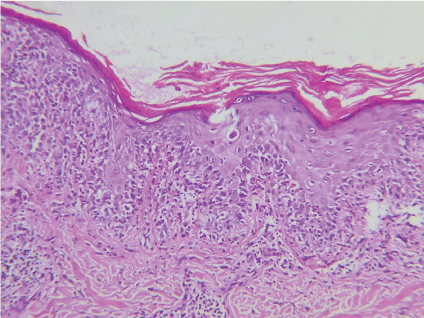 Figure 3: Necrotic keratinocytes and acantholysis. View Figure 3
Figure 3: Necrotic keratinocytes and acantholysis. View Figure 3
There may be intraepidermal cleft and acantholysis, or more rarely, subepidermal blisters. The clinical variants also have their respective histological features: (i) Pemphigus-Like: intra-epidermal cleft surrounded by mononuclear cells; ii) Bullous pemphigoid-like: subepidermal cleft with or without basal cellular vacuolization, and moderate mononuclear infiltrate in dermo-epidermal junctions; (iii) Erythema multiforme-like: dyskeratosis without cleft or with areas of epidermal separation, due to basal cell disintegration, and distinct perivascular infiltrate; (iv) Graft versus host disease-like: absence of epidermal separation, hyperkeratosis or hyperparakeratosis and dyskeratosis with or without vacuolar degeneration of basal cell layers and intense mononuclear interface dermatitis; (v) Lichen planus-like: hypergranulosis, dyskeratosis and lichenoid mononuclear infiltrate [3,19].
It is now accepted both autoantibody-mediated humoral immunity and cell-mediated immune mechanisms play a role in the development of PNP. The underlying immunological mechanism may shed light on the spectrum of clinical and histological features for a given case of PNP [20,21]. According to the dominant mechanism of cytotoxicity, pemphigus-like or LP-like features may be prominent. If the cytotoxicity is mediated by autoantibodies, a pemphigus-like clinicopathological presentation is prominent. In contrast, if the dominant mechanism is cell-mediated cytotoxicity, LP-like lesions may be prominent [22].
Direct Immunofluorescence (DIF) of a perilesional biopsy shows a typical pemphigus pattern with intercellular deposits of IgG/C3 in the epithelium and deposits of IgG/C3 with the same pattern in sweat gland ducts have recently been reported [23]. In addition, there may be linear deposits of IgG and/or C3 in the basement membrane zone, may reflect of the reactivity with BP230 [2]. Both patterns were seen in our patients. In patients 1 and 3, the results of DIF are negative and thus it is not a necessary diagnostic criterium for PNP [6-8].
In all forms of pemphigus Indirect Immunofluorescence (IIF) on monkey esophagus shows characteristic intercellular binding of IgG and often C3 in the epithelium. Given the strong expression of plakins, rat or monkey urothelium is the most appropriate substrate for screening for antibodies to PNP (Figure 4) [8,24]. Sera from patients with pemphigus vulgaris or foliaceus do not react with urothelium. If a specific detection system for autoantibodies to envoplakin or periplakin is not available, detection of anti-plakin-antibodies on IIF on rat or monkey bladder is necessary for diagnosis of PNP.
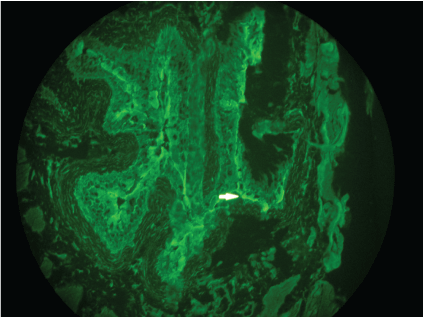 Figure 4: Indirect immunofluorescence using rat bladder epithelium as substance showing intercellular immunoglobulin G deposition. View Figure 4
Figure 4: Indirect immunofluorescence using rat bladder epithelium as substance showing intercellular immunoglobulin G deposition. View Figure 4
Autoantibodies to plakins are characteristic and diagnostic of PNP [2]. As structural components of desmosomal and hemidesmosomal plaques, their interaction with the keratin cytoskeleton of keratinocytes can result in the suprabasal clefting seen in PNP if affected by autoantibodies. The specific autoantibodies to plakins correlated with PNP include: desmoplakin I (250 kd), bullous pemphigoid antigen I (230 kd), desmoplakin II (210 kd), envoplakin (210 kd), periplakin (190 kd), plectin (500 kd), desmogleins (Dsgs) 1 and 3, and a 170-kd protein. This 170-kd protein has recently been identified as alpha-2-macroglobulin-like-1 (A2ML1), a broad range protease inhibitor expressed in stratified epithelia and other tissue damaged in PNP [25,26]. In 2016 a Japanese study indicated that epiplakin is one of the major PNP autoantigens and is related to PNP-related bronchiolitis obliterans [16]. A large cohort of 104 patients with PNP found statistically significant correlations between positive desmoglein 3 reactivity and genital lesions, and between positive desmoglein 3 reactivity and bronchiolitis obliterans [14].
Of those, the most specific for PNP is autoantibodies to envoplakin [27] and periplakin [28], followed by desmoplakin 1 and 2 [29], plectin [30], and a 170-kD protein [2]. Based on Joly and colleagues' study of 22 PNP patients [8], the presence of autoantibodies to periplakin or envoplakin was 82% sensitivity and 100% specificity. However, in a case reported by Sanz-Bueno J, et al., a patient with paraneoplastic autoimmune multiorgan syndrome secondary to a lymphoblastic T- cell lymphoma, had no detectable autoantibodies, who presented with a lichenoid dermatitis and vitiligo, later developing bronchiolitis obliterans and autoimmune hepatitis [31].
PNP is an autoimmune syndrome mostly seen in association with hematologic neoplasms including Castleman's disease [5,8,29,32-48], Non-Hodgkin's lymphoma [1,2,8,29,34,36,49-63], chronic lymphocytic leukemia [2,8,29,34,36,51,64-71], thymoma [1,2,5,8,29,34,46,66,72-77], Waldenstrom's macroglobulinemia [78-81], monoclonal gammopathy [8], Hodgkin's lymphoma [82], and centroblastic lymphoma [1]. Castleman's disease associated with paraneoplastic pemphigus is misdiagnosed frequently and easily in clinical practices. Furthermore, the mortality rate for this disease is very high. In a pediatric age group, paraneoplastic pemphigus has a striking association with Castleman disease and has a particularly poor prognosis in this age group [83].
The Nonhematologic neoplasms associated with PNP include follicular dendritic cell sarcoma [5,42,46,47,72,84,85], inflammatory myofibroblastic tumor [86], leiomyosarcoma [87], liposarcoma [88], malignant nerve sheath tumor [89], inflammatory fibrosarcoma [90], reticulum cell sarcoma [91], and malignant melanoma [92], squamous cell carcinoma of the vagina [93], and adenocarcinoma of the colon [55], prostate [94,95], pancreas [88,93], breast [36], and lung [96,97] (Table 1). Follicular Dendritic Cell Sarcoma (FDCS) is a rare tumor associated with paraneoplastic pemphigus. It is an uncommon neoplastic proliferation of spindled to ovoid cells with morphologic and immunophenotypic features similar to normal follicular dendritic cells. While most follicular dendritic cell sarcomas arise from lymph nodes, at least one-third occur in extranodal sites. Inflammatory Myofibroblastic Tumour (IMT) is a rare low-grade sarcoma of fibroblasts and myofibroblasts associated with inflammatory cells, most commonly occurring in the lung. In a study [98], a case of PNP associated with IMT of the mediastinum was reported and the patient had a favourable outcome following surgical resection and treatment with a systemic steroid, azathioprine, and i.v. immunoglobulin.
Table 1: Paraneoplastic pemphigus: associated neoplasms. View Table 1
In 1990, Anhalt initially proposed five criteria for the definition of a PNP case [2]: (1) Painful mucosal erosions and polymorphous skin eruption in the context of a neoplasia; (2) Histological changes (acantholysis, keratinocyte necrosis, interface dermatitis); (3) DIF showing IgG and complement deposition in intercellular substance and basement membrane zone; (4) IIF with the same deposition as for DIF, in skin, mucosa and simple, columnar, and transitional epithelium and (5) Demonstration of serum antibodies through immunoprecipitation of a complex of four keratinocyte proteins (250, 230, 210 e 190 kd). In 2004, Anhalt proposed minimal diagnostic criteria for PNP [29]. (1) Clinical: painful progressive stomatitis with preferential involvement of tongue; (2) Histological: acantholysis or interface dermatitis; (3) Immunological: presence of antiplakin antibodies (at least periplakin and envoplakin). The pivotal criterium of PNP is autoantibodies directed against desmosomal plakin proteins: desmoplakin I (250 kDa), desmoplakin II (210 kDa), envoplakin (210 kDa), periplakin (190 kDa), and α2macroglobulin-like-1 protein (170 kDa). In addition, autoantibodies against Dsg-1, Dsg-3, plectin and 230 kDa bullous pemphigoid antigen can be detected. These antiplakin antibodies should be revealed by immunoprecipitation or immunoblotting, in addition to positive IIF in monkey esophagus and rat bladder. Anti-Dsg-3 ELISA may also be positive, but this does not discriminate between PNP and other pemphigus variants (PV and PF). (4) Association with lymphoproliferative disorder: Non-Hodgkin's lymphoma and chronic lymphocytic leukemia generally in cases with previous diagnosis (2/3 of cases), and Castleman's disease, abdominal lymphoma, thymomas or retroperitoneal sarcoma in cases with ocult neoplasia at the time of diagnosis of PNP (1/3 of cases) [3].
Patients with a diagnosis of PNP without previous diagnosis of a neoplasia - about 17% of PNP cases - must be investigated with complete blood count with differential leukocyte, serum protein electrophoresis, computerized tomography (chest, abdomen, and pelvis), and biopsies of bone marrow, lymph nodes, or solid tumor, according to indication [3].
The prognosis of PNP is generally poor, and the disease is often fatal. The mortality of patients with PNP is 75% to 90% [3]. However, a recent study conducted in France [99] revealed a mortality of 51%, 59% and 69% in 1, 2 and 5 years, respectively.
The treatment of PNP to date has been rather disappointing. The skin and mucosal eruptions cause much morbidity and are a frequent cause of death rather than the malignancy itself [100]. The response to therapy is generally poor. In the first place, therapy is aimed at curtailing the production of autoantibodies [101-103]. The proven efficacy therapies include Cyclosporine A [104], Rituximab [1,13,58], Cyclophosphamide [1,67], Mycophenolate mofetil [68]. The most widely suggested specific treatment combines prednisone (0.5-1.0 mg/kg) with cyclosporine (5 mg/kg), and may also include cyclophosphamide (2 mg/kg) [3]. Initially, high-dose corticosteroids form the first line of defense, and are effective to some extent [100]. The addition of steroid-sparing agents, such as azathioprine [100], cyclosporine A [104], and mycophenolate mofetil [68], may reduce steroid intake and thus limit side-effects; however, a high level of caution is advised in patients with paraneoplastic pemphigus and confirmed malignancy, where immunosuppression is paramount and dictates the mainstay of treatment options. Rituximab may be indicated, especially because of association with non-Hodgkin's lymphoma, though there are reports of complications and low therapeutic response [3].
A better prognosis can be expected if the malignancy is less aggressive. This is the case with Castleman's disease and thymoma [7,46]. Early detection and resection of the tumor are essential for the treatment of the patients with castleman's disease. The associated tumors should be resected completely. During the operation, it is important to block the blood supply of the tumor first and to avoid squeezing the tumor during operation. Complete resection is prerequisite for a good prognosis. Using i.v. immunoglobulin before, during and after the operation in a total dose of 1-2 g/kg is recommended to block the circulating autoantibodies released from the tumor, which could cause pulmonary damages [7]. However, except in cases associated to Castleman's disease, treatment of neoplasia is not associated with improvement of PNP in general.
Respiratory failure due to bronchiolitis obliterans is one of the most important causes of death in patients with PNP/PAMS. Pulmonary disease is irreversible and tumor resection or complete response to neoplasia treatment does not alter the progression of respiratory disease [3].
None.
None to declare.
None.