Congenital anosmia (CA) is rare comparatively to acquired anosmia. The pathophysiology and the genetic background remain poorly understood. This study aims to review the main clinical and Magnetic Resonance Imaging (MRI) findings to discuss the diagnostic strategy, the pathophysiology and the genetic basis of the different forms of CA.
A series of 60 patients with congenital anosmia was reviewed retrospectively out of 425 non-sinus-related anosmia and compared to the literature data. CA was assessed by Ear Nose Throat (ENT) workup, psychophysical olfactometry and MRI evaluation of olfactory bulbs (OB) and central olfactory structures.
The 60 cases of CA include 16 isolated congenital anosmia and 44 syndromic congenital anosmia especially 20 Bardet Biedl syndrome and 22 Kallmann syndrome. The diagnosis of CA is often overlooked or delayed especially for isolated CA with no or few complaints about olfaction impairment compared to acquired anosmia. MRI shows in most cases hypoplasia or aplasia of OB with a strong correlation between global OB visual evaluation and OB volumetric evaluation for trained radiologists and often central olfactory abnormalities needing 3D MRI imaging.
The pathophysiology involving peripheral and/or central olfactory abnormalities and the involved genetic mechanisms remain poorly understood. Complete genomes sequencing in affected patients and families with well delineated CA (ENT workup, olfactometry, MRI) and studies of different CA animal models constitute a challenge for better understanding of this rare and heterogeneous group of CA.
Anosmia, Congenital anosmia, Bardet Biedl syndrome, Kallmann syndrome, MRI, Olfactory bulb
CA: Congenital Anosmia; MRI: Magnetic Resonance Imaging; ENT: Ear, Nose and Throat; OB: Olfactory Bulb; ICA: Isolated Congenital Anosmia; SCA: Syndromic Congenital Anosmia; BBS: Bardet Biedl Syndrome; KS: Kallmann Syndrome; STOE: Suprathreshold Evaluation of Olfaction
Loss of smell (anosmia) is common in the general population (prevalence about 5%) and is associated with reduced quality of life [1]. The etiologies are numerous and various, and the prevalence increases with age. Patients with congenital anosmia (CA) or congenital olfactory dysfunction have no memory of ever being able to smell. CA remains rare (1% of the anosmia population) and can be isolated (Isolated Congenital Anosmia, ICA) or associated with various diseases (Syndromic Congenital Anosmia, SCA) like Bardet Biedl syndrome (BBS) and Kallmann syndrome (KS) [1]. The pathophysiology and the genetic basis are still poorly understood especially for non-syndromic CA. Complete study of olfactory bulbs, sulcus and olfactory central structures with 3D MRI imaging and complete sequencing of the genomes of CA patients and CA families constitute a challenge for better understanding for these different forms of CA.
This review aims to describe the different forms of CA, the diagnosis difficulties, the olfactory evaluation with different technics and the main clinical and MRI findings (evaluation of olfactory bulb (OB) and central olfactory structure by 3D MRI) in a series of 60 cases of CA [2,3].
This study for BBS patients received ethical approval for the study PHRC national Bardet Biedl IDRCB 2007-A00868-45.
For this retrospective single center study all patients were evaluated by the same Ear Nose and Throat (ENT) specialist over a period of 30 years through a university department of rhinology and a private ENT office. Out of a series of 425 patients with non-sinonasal-related anosmia especially post-infectious (56%), posttraumatic (14%) and anosmia associated with toxins/medications, neurologic diseases and idiopathic anosmia (16%), 60 CA patients (14%) were included for this study.
A senior ENT specialist evaluated all CA according to the following protocol: (i) Clinical evaluation of olfaction and its dysfunction: history taking, onset, duration, fluctuation, other nasal associated symptoms, past medical history looking for head trauma, respiratory infections, sinonasal surgery, neurosurgery, toxins/medications, occupational exposure, family history of CA, non-nasal associated symptoms (ii) ENT examination with nasal endoscopy (iii) Olfaction evaluation using one or two different psychophysical methods namely a suprathreshold evaluation of olfaction (STOE) and/or the UPSIT French version (Sensonics, Pennsylvania NJ), (iv) MRI for nasal cavities, sinuses, OB and quantitative evaluation of the morphological central olfactory structures alterations (Voxel-Based Morphometry) when possible or qualitative evaluation of overall brain morphology (qualitative assessment of cortical atrophy, anomalies of the central structures), (v) Evaluation of the non-nasal associated symptoms by workup of different specialists (pediatrician, endocrinologist, urologist, gynecologist, ophthalmologist, geneticist) according to the associated clinical symptoms, (vi) Genetic analysis for all BBS patients and for six KS patients.
BBS is a well-recognized ciliopathy with early onset retinitis pigmentosa, polydactyly, obesity, hypogonadism, renal and cognitive impairment, congenital or early in life dysosmia with genetic background [3]. BBS genes have been screened thanks to high throughput sequencing using a custom panel targeting depending on the version 31 genes or 58 genes including the 19 BBS genes and other related pathologies such as Alstrom syndrome, Meckel syndrome, nephronophthisis... [4].
KS diagnosis was performed according to the criteria of the European Consensus Statement [5]. The association of congenital hypogonadotropic hypogonadism (CHH) with absence of puberty and infertility associated with anosmia or hyposmia is termed KS (50% of patients with CHH). More than 25 causal genes have been described to date [5]. Only in 6 KS cases molecular diagnosis has been performed with detailed phenotype analysis and sequencing of the coding sequences of KAL1, FGFR1, FGF8, PROKR2 and PROK2 [6].
Olfaction evaluation was performed on the basis provided by previous work [2,3]. The suprathreshold evaluation was based on the perception and identification of 10 odors in three trials with randomized order (53 patients) [3]. For the UPSIT evaluation, identification of the odors was made using the technique of multiple forced choices of 40 odors (37 patients) [7]. There was a strong correlation between the two tests [2,3].
For this study MRI evaluation was not available for all patients especially for the first included patients because access to MRI was limited at that time. Some rare patients refused MRI "being without any utility in the absence of treatment for CA" and in other patients MRI was focused on the pituitary gland especially by suspicion for KS and did not retrospectively allow a precise and complete study of OB nor- central olfactory structures needing 3D MRI imaging.
Images were acquired on 1.5T (20 cases) or 3T (27 cases) MRI unit. MRI protocols that included coronal T2-weighted sequence covering the nasal cavities allowed evaluation of olfactory bulb and sulcus without real difference between 1, 5T and 3T MRI. MRI protocols that included whole-brain imaging allowed qualitative evaluation of cortical atrophy and central structures. 3T MRI protocols that included T1-weighted SPACE (Sampling Perfection with Application optimized Contrasts using different flip angle Evolution) sequence with 1-mm slices allowed quantitative evaluation of the morphological central olfactory structures alterations.
Finally, CA patients have been identified by using the following working definition. For the 60 CA the past history found no head injuries, no respiratory infections and no other causative condition of anosmia. Clinical evaluation, nasal endoscopy, MRI evaluation did not find any incidental nasal-or sinus-related medical or surgical conditions or disorders explaining anosmia. Anosmia or severe hyposmia was associated with no memory of ever been able to smell.
The series of CA included 31 male and 29 female patients with a mean age of 27.7 years at time of diagnosis (extreme ages: 8-61 years). The 60 CA included 16 ICA (7 male, 9 female, mean age: 36.7 years) and 44 SCA with 20 BBS (13 male, 7 female, mean age: 28.1 years), 22 KS (10 male, 12 female, mean age: 21.9 years) (Table 1, Table 2 and Table 3), one CHARGE syndrome in a male of 20 years and one rare association of CA with atrophic rhinitis, bronchiectasis and common variable immunodeficiency in a young child of 12 years.
Table 1: Isolated congenital anosmia. View Table 1
Table 2: BBS patients and OB visual evaluation according to Braun, et al. [2]. View Table 2
Table 3: Syndromic congenital anosmia and Kallmann syndrome. View Table 3
In ICA there were no or only few complaints about alteration of quality of life like cognitive impairment, social life alteration, loss of appetite, depression, anxiety compared to acquired anosmia (Table 1). These complaints and symptoms were evaluated routinely in all CA subjects.
Dysosmia was a constant and cardinal feature in our series of 20 BBS patients according to UPSIT, STOE and MRI (OB and central olfactory structures) evaluation for this ciliopathy associated anosmia (Table 2).
In 22 patients CA was associated with hypogonadotropic hypogonadism in a KS [5] (Table 3).
In one case a rare nasobronchial dystrophy associated CA, severe early in life non ozaenic atrophic rhinitis, bronchiectasis needing lobectomy (lingula) and common variable immunodeficiency (CVID) (especially IgG deficit).
In one case CHARGE syndrome associated CA, coloboma, incomplete choanal atresia, heart abnormalities, growth retardation, genital and urinary abnormalities and hearing loss.
Among the 16 ICA with complete anosmia in all cases, 10 patients had MRI olfactory bulb global visual evaluation performed blindly and separately by two experienced radiologists (SR and SM) showing bilateral OB aplasia in 7 cases, OB hypoplasia in 1 case and normal OB in 2 cases. CT-scan of sinuses and nasal cavities performed in 9 cases was normal (Table 1, Figure 1 and Figure 2).
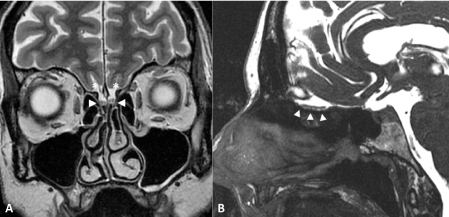 Figure 1: Normal olfactory bulbs.
Figure 1: Normal olfactory bulbs.
MRI of normal-size olfactory bulbs (arrowheads) on coronal T2-weighted imaging (A) and sagittal Fast Imaging Employing Steady-state Acquisition, FIESTA (B). Olfactory sulci are well seen (star). View Figure 1
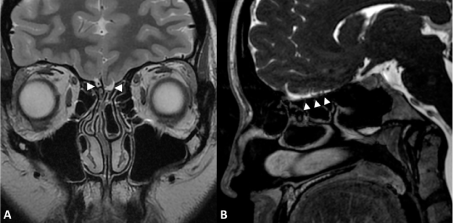 Figure 2: Patient 10 with Isolated Congenital Anosmia.
Figure 2: Patient 10 with Isolated Congenital Anosmia.
MRI of aplastic olfactory bulbs on coronal T2-weighted imaging (A) and sagittal Fast Imaging Employing Steady-state Acquisition, FIESTA (B). The olfactory fossae (arrowheads) does not contain any olfactory bulb. Right olfactory fossa contains a small artery. Olfactory sulci are not visualized. View Figure 2
For 20 BBS patients, OB visual evaluation was abnormal in 18 out of 20 cases for the first radiologist (SR) and in 20 out of 20 cases for the second radiologist (SM) comparatively to a series of 14 healthy volunteers with normal OB as reported in detail in a previous publication [2] (Table 2).
In 22 KS patients with anosmia in all cases (UPSIT and/or STOE), 17 MRI were available for both pituitary gland evaluation comparatively to hormonal evaluation and for OB evaluation. The results are summarized in Table 3.
In the syndromic association of CA, atrophic rhinitis, bronchiectasis and CVID (Figure 3) and in the CHARGE syndrome MRI showed complete aplasia of the OB. These two patients had complete anosmia.
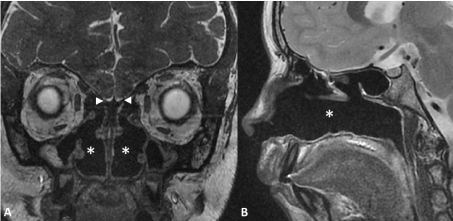 Figure 3: Congenital anosmia and atrophic rhinitis in a 12-years-old child.
Figure 3: Congenital anosmia and atrophic rhinitis in a 12-years-old child.
MRI of aplastic olfactory bulbs associated with severe atrophic rhinitis on coronal Fast Imaging Employing Steady-state Acquisition, FIESTA (A) and sagittal T2-weighted imaging (B). The olfactory fossae (arrowheads) does not contain any olfactory bulb. The other major finding is empty nasal cavities (star) with atrophic turbinates and hypoplasic paranasal sinuses. View Figure 3
In 5 cases of CA (2 ICA, 3 KS) OB were considered normal by both radiologists.
Central olfactory structures could be evaluated quantitatively in all 20 cases of BBS and qualitatively in 3 cases of ICA and in 4 cases of SCA. In 20 BBS patients MRI highlighted correlation between gray mater volume reduction in several brain areas and olfaction dysfunction [2,3]. In 3 -ICA patients and 4 -KS patients qualitative evaluation of brain morphology showed mild volume loss in the temporal and frontal lobes.
All patients have not been genetically evaluated especially ICA patients. In the 20 BBS patients the mutated BBS gene has been identified: BBS 1 to 19 (Table 2). In 6 recent cases of KS with aplasia of OB a molecular diagnosis has been performed and involved KAL1 in 4 cases including 2 brothers, a heterozygous mutation in gene FGFR1 and PROKR2 in one case and the results were awaited at the time of writing in one case (Table 3). For the patient with atrophic rhinitis, bronchiectasis and CIVD, genome sequencing was not contributive.
CA is rare compared to the high frequency of acquired anosmia and of age-related olfactory impairment. The diagnosis remains difficult and is often delayed considering the few complaints mentioned spontaneously especially by patients presenting ICA and the difficulties to evaluate the olfactory dysfunction. In this series, the mean age at diagnosis was 36.7 years for ICA compared to 21.9 years for KS and 28.1 years for BBS.
In SCA, anosmia is a part of a large spectrum of diseases e.g- KS, different ciliopathies like BBS [2,3], Alström syndrome, Leber Congenital Amaurosis (LCA) [8] and other rare genetic disorders like Refsum disease, Usher syndrome, CHARGE syndrome, Johnson Mc Millin syndrome and congenital insensitivity to pain [9]. In our series of KS patients, the 21 patients who had an olfactory test (UPSIT and/or STOE) had anosmia. These results are similar to those of Ottaviano, et al. [10] who found 94.7% of anosmia and 5.3% of hyposmia in a cohort of 38 KS patients. Another study on 21 patients found different results: 66.6% of anosmia and 33.3% of hyposmia [11]. Our BBS patients obtained similar results for olfactory tests with the UPSIT test compared to another cohort of 17 BBS patients [12]. In this cohort, no patients had MRIs. Of the 17 patients, none had a normal UPSIT test like our patients. Their patients had mild anosmia in 35.3% of cases vs. 30% for our patients, severe anosmia in 29.4% of cases vs. 30% and an anosmia in 35.3% vs. 40%.
In clinical practice only subjective and not objective olfactory tests are usually used for diagnosis of dysosmia needing cooperation of the subject. The results are deeply related to the olfactory education and memory of the patients. Therefore, MRI becoming more common in the last decade is useful for diagnosis of CA or early in life anosmia. In most cases, MRI shows aplasia or hypoplasia of OB even if diagnosis of CA is often based only on confirmation of lack of smell and history of absent smell from birth [2,3,9,13,14]. OB hypoplasia/aplasia is the most frequent or well-known MRI finding related to anosmia independently of its origin and MRI evaluation of the olfactory sulcus (OS) (depth and form) is a good indicator for isolated anosmia [15]. For trained radiologists there is a strong correlation between global OB visual evaluation and OB volumetric evaluation for evaluation olfactory dysfunction [2], which means that global OB visual evaluation is an accurate and objective criterion for diagnosis of anosmia and for confirmation of the results of the psychophysical olfactory tests. Olfactory self-assessment is unreliable and even absent OB with MRI is not an absolute proof of anosmia [16,17].
For Huart, et al. [15] and Hummel, et al. [18], the evaluation of the olfactory sulcus (depth and form) is a useful clinical indicator for isolated anosmia (IA). In fact, a depth of olfactory sulcus less than 8 mm clearly indicates IA. In our series, this evaluation of the olfactory sulcus was not possible for all CA cases because of the old data for some available MR scans allowing no statistical conclusions. We have consequently focused our imaging study on the OB and the central olfactory structures abnormalities. For BBS patients OB were abnormal in 18 out of 20 cases (radiologist SR) and in 20 out of 20 cases (radiologist SM). OB were abnormal in 14 out of 17 KS cases (82.4%) and in 8 out of 10 ICA cases for both radiologists. For MRI in BBS patients, there is no other study as the one published by Braun, et al. [3]. Many studies use MRI to investigate the presence of OBs and OS anomalies in KS. Koenigkam-Santos, et al. [11] found a similar percentage of abnormalities (85%) in the MRI of olfactory pathways, including OB aplasia/hypoplasia and/or abnormal OS. Ottaviano, et al. [10] showed that there was a significant difference in mean OB volumes between KS patients and controls, and that anosmic had significantly smaller OBs compared to hyposmic patients whereas hyposmic patients did not differ significantly from controls.
Nevertheless in 2 cases out of the 10 ICA with MRI and in 3 other cases out of the 17 KS with MRI, OB visual evaluation was normal. We have not found olfactory phenotypic spectrum reported by Lewkowitz-Shpuntoff, et al. [19] for KS patients. Apparently normal aspect and volume of OB has been described both in ICA [20] and KS [21,22] raising the possibility of molecular abnormalities of the olfactory epithelium or of functional defect in olfactory cortex without certainty about primary congenital origin or secondary origin to neuronal defect for these patients with CA and normal OB.
On the other hand, MRI is useful for differential diagnosis of CA: meningioma, post-traumatic injury, sinusal masses.
For Yousem, et al. [23] congenital anosmia appears to be an olfactory bulb-olfactory tract phenomenon rather than a cerebral process with only inconstant or mild temporal and/or frontal lobe volume loss for KS. Nevertheless, another study of dysosmia in BBS patients found in several brains areas central olfactory structures alterations of probably multifactorial origin (Voxel Based morphometry) [3]. Similar findings are reported in some cases of CA [18]. In other studies of ICA and SCA MRI shows associated central olfactory structures alterations e.g- atrophied and hypertrophied brain (Grey matter/White matter) areas in BBS [3] and in CA [23,24]. This quantitative evaluation of central olfactory structures with voxel-based morphometry (VBM) remains rarely performed in the literature for CA study [25-28]. There is a need for further investigations to study the correlations between olfaction impairment and central brain alterations and the causal relationship between olfactory dysfunction and neuroanatomical differences taking into account the possible role of confounders especially in SCA [3].
Concerning the olfactory epithelium, the results of epithelium biopsies still remain debated, given sampling errors taking into account the difficulties of olfactory biopsies in human, but may show absence or abnormalities of the olfactory epithelium in CA patients [9,23].
The pathophysiology of CA can be focused on peripheral and/or central alterations of olfactory structures: (i) Abnormal olfactory clefts and bilateral choanal atresia like in CHARGE syndrome [29], (ii) Abnormal olfactory mucosa (olfactory cells and/or olfactory receptors) in BBS patients, LCA, Alström syndrome and in mice models like BBS1, BBS4 et BBS8-null mice [30-32], (iii) Abnormal OB (aplasia or hypoplasia) [2,13,14], (iv) Alterations or modifications of central olfactory brain structures (Grey Matter, White Matter of several brain areas by Voxel-Based Morphometry [3,24], (v) Abnormal embryologic neuronal migration process from the nasal placode in KS with anosmia and hypogonadotropic hypogonadism [5,33]. In a rare case, CA can be a part of a complex rhinobronchial dystrophy with aplasia of the OB, bronchiectasis, severe atrophic rhinitis and DICV without a clear link between these different symptoms.
Attempts to identify genetic mutations linked to CA confirm the heterogeneity of the genetic transmission (dominant or recessive inheritance) with multiple genes candidates, various microdeletions and duplications after sequencing of the genomes in affected patients and families. On the other hand, for genetic studies it must be kept in mind the difficulties to objectively test dysosmia, to formerly prove its congenital nature, to understand its pathophysiology and to prove its genetic origin which is often heterogeneous like anosmia itself. Different genes involved in CA have been reported in the literature.
Cases of ICA associated with life-long inability to smell without other associated symptoms are often sporadic. In published cases with family history of ICA the genetic transmission seems to be autosomal dominant inheritance with reduced penetrance in some cases and with linkage on chromosome 18 in some cases without disease-causing mutations by direct sequencing and with significant linkage to a 45cM region on chromosome 4 for another study [9]. In a recent study of five affected family members the genetic findings provide support for role of CNGA2 gene with pathogenicity in ICA in humans and in CNGA2 knockout mice model congenitally anosmic [34]. With whole-exome sequencing technic in a CA family, a rare X-linked missense mutation in the TENM1 (terneurin) gene was identified as molecular genetic basis for human and also for a TENM1 mice model [35].
The inheritance of KS of embryologic origin seems to be heterogeneous, with dominant autosomic, recessive autosomic and linked to X chromosome inheritance. More than 25 genes (KAL1, FGFR1, FGF8, CHD7, PROK2 and PROKR2) have been reported in the literature and seem to explain about 30% of KS (5,6,9). CHARGE syndrome has been linked to a CDH7 mutation also involved in KS [36]. In some genetic ciliopathies with retinal dystrophy like BBS, Alström syndrome, LCA and also in Refsum disease and Usher syndrome anosmia has been reported [5,7-9,16,17,37,38]. For instance, olfactory impairments have been reported in BBS patients and also in KO mice deleted for the BBS1, BBS4 et BBS8 genes with various disorders in olfactory epithelium (olfactory cells/receptors) [30-32]. Loss-of- function of the cilia-centrosomal protein CEP290/NPHP6 is associated with Joubert and Meckel syndrome and a hypomorphic CEP290 mutation with LCA [39]. Anosmia in congenital insensitivity to pain has been attached to SCN9A mutations with failure of synaptic signaling at the terminal axon providing a link between pain and olfactory modalities [40].
Despite significant genetic heterogeneity, 20 BBS (100%) patients were genetically characterized (BBS 1 to 19) and also 6 KS (27%) patients evaluated by genome sequencing. Unfortunately, some KS patients and ICA patients have not been genetically evaluated. Nevertheless the role of the numerous candidate genes for CA and their gene application in the pathophysiology remain unclear especially for ICA [9].
Our study of 60 CA confirms the diagnosis difficulties for these rare CA classified in ICA and SCA. It highlights the interest of MRI imaging (i) OB visual and volumetric evaluations with a good correlation for trained radiologists [2,3], (ii) Olfactory sulcus depth as indicator for isolated anosmia [15] and (iii) Quantitative evaluation of central olfactory structures with VBM performed only in few studies especially for BBS patients with congenital dysosmia which constitute a cardinal feature in BBS patients [3]. Nevertheless, there is a lack of correlation between OB MRI evaluation and dysosmia in some rare cases of CA with normal OB MRI imaging and anosmia. The physiopathology and the involved genetic mechanisms remain poorly understood especially for ICA.
Finally, the main difference between syndromic and non-syndromic anosmia is linked to associated symptoms (BBS, KS) and to abnormal embryologic migration process for KS. The characteristics of anosmia itself remain similar. Comparison between olfactory deficit globally and for specifics scents may be interesting but has not been performed in our study nor in our literature review.
Limitations of our study may be, despite the large series of CA, the retrospective nature of this study, the absence of complete MRI evaluation of OB, olfactory sulcus and central olfactory structures with VBM and the lack of genetic testing for all patients.
Patients suffering from CA live in a world without olfactory dimension since birth or early in life [41]. Nevertheless, their complaints are less evident than for acquired anosmia by adapting strategies to the olfactory deficit in daily life [42]. This congenital disorder in isolated or syndromic form is often overlooked or diagnosed with delay especially for ICA. The pathophysiology of CA remains poorly understood with peripheral and/or central olfactory abnormalities which begin to be better known by MRI studies (standard MRI, functional MRI, 3D MRI, Voxel-Based Morphometry), studies of CA animal models and biopsies of olfactory epithelium in CA patients. In our study, 100% of the BBS cases have been genetically characterized and only 27% of KS patients, which represents 43% of all our patients.
Complete sequencing of the genomes of affected patients and families with well-defined CA [43] constitutes a challenge for our knowledge about the genetic pathways and the molecular mechanisms linked to this heterogeneous group of CA.
Jean Muller, MD.
All authors have contributed to the production of this article.
No sources of support.