Hypertrophic cardiomyopathy (HCM) is a disorder characterized by ventricular hypertrophy and myocyte disarray that increases the risk of arrhythmic sudden cardiac death. HCM phenocopies are disease entities sharing a left ventricular hypertrophy phenotype but arising from a different pathologic mechanism. Patients with PRKAG2 syndrome (PS), a glycogen storage disease, are often misdiagnosed as having HCM. Clinicians must be familiar with red flags alerting to the possibility of this important disorder. Genetic testing allows recognition of PS and other HCM phenocopies, facilitates targeted therapy and screening, and helps identify relatives carrying pathogenic variants. Potential treatment investigations include SGLT2 inhibitors and alglucosidase alfa replacement.
HCM: Hypertrophic Cardiomyopathy; PS: PRKAG2 Syndrome; WPW: Wolff-Parkinson-White; NT-proBNP: N-Terminal Pro-Brain Natriuretic Peptide; ECG: Electrocardiogram; NSVT: Non-Sustained Ventricular Tachycardia; ICD: Implantable Cardioverter-Defibrillator; AMPK: AMP-Activated Protein Kinase; SCD: Sudden Cardiac Death; GLUT4: Glucose Transporter Type 4; G-6P: Glucose 6-Phosphate; CMR: Cardiac Magnetic Resonance; LGE: Late Gadolinium Enhancement; SGLT2: Sodium-Glucose Linked Transporter 2
We report a case of a 48-year-old man with a past medical history of “hypertrophic cardiomyopathy”, complete heart block, and heart failure with preserved ejection fraction who was seen in the clinic after a hospital discharge. He reported a family history of Wolff-Parkinson-White (WPW) from his maternal side, and two healthy daughters. At the age of 24 years, he developed a complete heart block which required the implantation of a permanent pacemaker. Due to his young age, he underwent genetic testing, however he was told it was unrevealing.
During his mid-forties he developed progressive shortness of breath, palpitations, and paroxysmal nocturnal dyspnea, which led to hospital admission. He was diagnosed with decompensated congestive heart failure and was treated with intravenous diuretics for several days.
High-sensitive troponin ranged from 70-115 pg/mL, and the N-terminal pro-brain natriuretic peptide (NT-proBNP) was 1,083 pg/mL (< 100 pg/mL), the kidney function and hematology panels were normal. The 12-lead electrocardiogram (ECG) showed a ventricular paced rhythm. A heart catheterization demonstrated a large left anterior descending artery, which gave rise to prominent septal perforator branches and no obstructive coronary artery disease. A two-dimensional transthoracic echocardiogram (Figure 1) showed severe concentric hypertrophy, grade III diastolic dysfunction and preserved left ventricular systolic function. A treadmill stress test performed previously had shown no evidence of arrhythmias, ischemia, or hypotension. Interrogation of his device showed a few episodes of non-sustained ventricular tachycardia (NSVT).
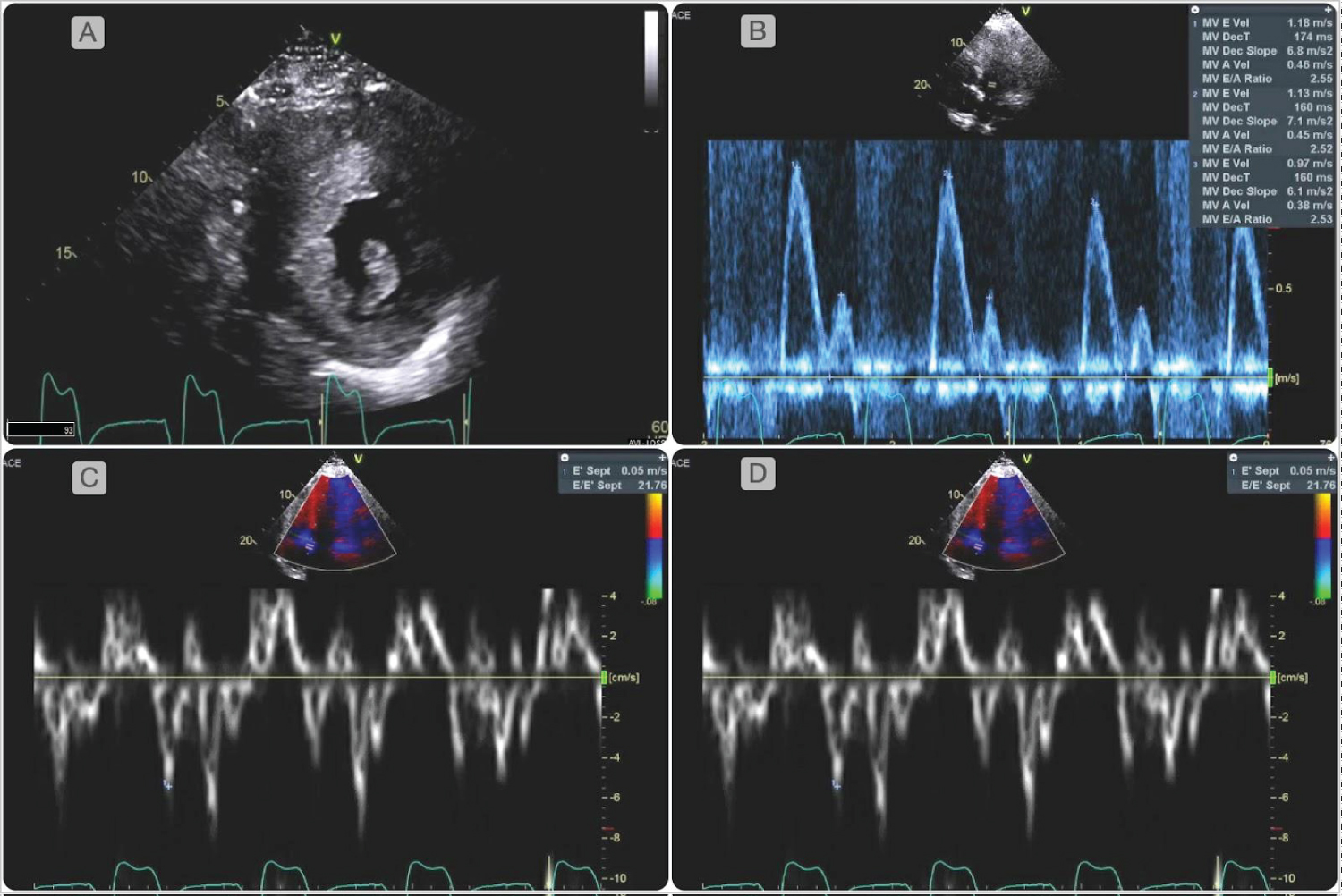 Figure 1: Two-dimensional transthoracic echocardiogram showed mildly increased left atrial volume index (38 mL/m2). Diastology parameters were abnormal as evidenced by an increased ratio between early mitral inflow velocity and the average mitral annular early diastolic velocity (E/e' 24), reduced mitral valve medial and lateral annular velocities (medial e' 5 cm/second, lateral e' 4 cm/second), consistent with grade III diastolic dysfunction. There was severe concentric hypertrophy with a left ventricular mass index of 423 g/m2, and a relative wall thickness of 0.55. There was no evidence of left ventricular outflow obstruction, systolic anterior motion of the mitral valve or apical aneurysms.
View Figure 1
Figure 1: Two-dimensional transthoracic echocardiogram showed mildly increased left atrial volume index (38 mL/m2). Diastology parameters were abnormal as evidenced by an increased ratio between early mitral inflow velocity and the average mitral annular early diastolic velocity (E/e' 24), reduced mitral valve medial and lateral annular velocities (medial e' 5 cm/second, lateral e' 4 cm/second), consistent with grade III diastolic dysfunction. There was severe concentric hypertrophy with a left ventricular mass index of 423 g/m2, and a relative wall thickness of 0.55. There was no evidence of left ventricular outflow obstruction, systolic anterior motion of the mitral valve or apical aneurysms.
View Figure 1
Genetic testing was performed using the Invitae panels for Arrhythmia and Hypertrophic Cardiomyopathy (Invitae, San Francisco, California). A pathogenic variant of the PRKAG2 syndrome was identified (c.905>A p.Arg302Gln). Additionally, two variants of uncertain significance were found of the JUP and PDLIM3 genes (c.529C>T and c.278C>G).
Arrangements were made for the management of heart failure, longitudinal monitoring of disease progression and arrhythmic burden via serial imaging and biomarker measurements with pacemaker interrogation. In light of his episodes of NSVT he was referred to an electrophysiologist for the consideration of an implantable cardioverter-defibrillator (ICD) implantation.
Although our patient carried the diagnosis of HCM, the presence of complete heart block at an early age, severe concentric hypertrophy, along with his family history of WPW, hinted at the possibility of an undiagnosed inherited cardiomyopathy. Genetic testing helped establish the diagnosis by identifying a pathogenic variant c.905>A p.Arg302G in the AMP-activated protein kinase (AMPK) gene related to the PS.
PS is hallmarked by the triad of cardiac hypertrophy, advanced atrioventricular block, and ventricular pre-excitation; however, it often goes undiagnosed as cardiac hypertrophy may be attributed to HCM [1]. Whereas myocyte disarray, and sarcomeric protein mutations are present in HCM, PS is driven by an entirely different mechanism involving excessive glycogen accumulation in the myocardium. Distinguishing PS from sarcomeric HCM is of utmost importance due to its poorer prognosis [2].
The genetic basis of PS was discovered in 2001, when a specific genetic variant was described in a family of patients with WPW. Common disease-causing variants include c.905>A (Arg302Gln) and c.1463A>T (Asn488Ile). Inheritance occurs in an autosomal dominant fashion with a penetrance of nearly 100%, and up to 44% of those affected experience sudden cardiac death by the age of 40 [2]. As demonstrated in the largest PS cohort to date, patients may not exhibit all three of PS classical features. Lopez-Sains, et al. analyzed outcomes from the largest cohort of PS cases (n = 90) and found that 33% of patients did not have pre-excitation, and 29% of patients had atrial fibrillation [3]. When compared to a less common variant, our patient’s variant has been associated with a higher propensity for pacemaker placement, pre-excitation, and sudden cardiac death (SCD) [1].
The gene implicated in PS encodes for the regulatory gamma 2 subunit of AMPK [3], an intracellular energy-sensing enzyme. Pathogenic variants in this gene lead to chronic activation of AMPK, which result in an increased expression of glucose transporter type 4 (GLUT4) transporters allowing higher amounts of glucose to enter the cell, and thus intracellular levels of glucose 6-phosphate (G-6P) rise. In turn, G-6P allosterically activates Glycogen synthase causing the formation of the excessive and unnecessary amounts of glycogen demonstrated in pathologic specimens (Figure 2) [4,5].
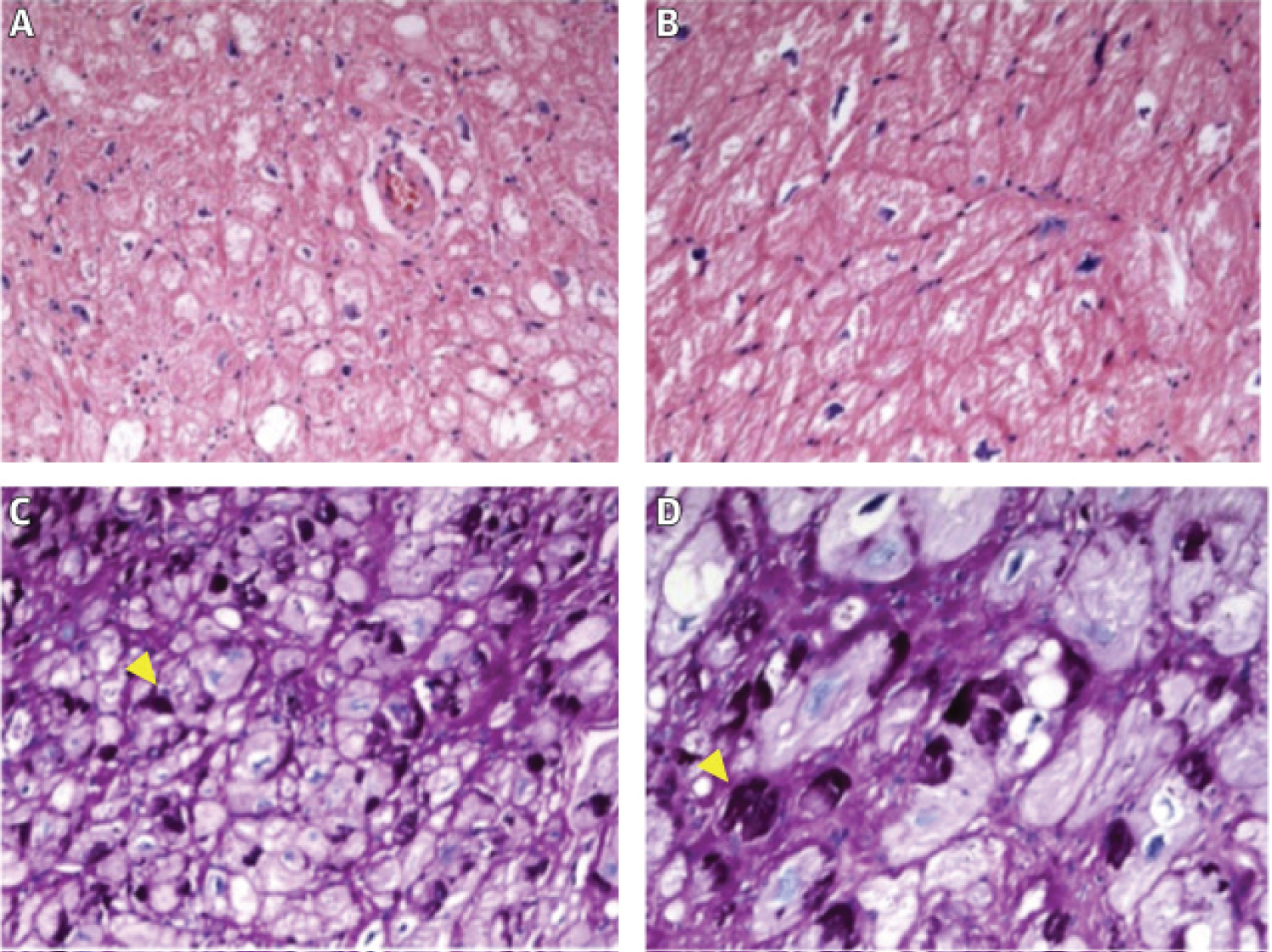 Figure 2: Typical findings in PRKAG2 syndrome. (A,B) Hematoxylin-eosin staining displaying hypertrophied myocytes. Magnification 200× and 400×; (C,D) Periodic acid Schiff (PAS) staining positive for glycogen accumulation in cardiomyocyte vacuoles. Magnification 200× and 400×. Yellow arrowheads indicate PAS+ deposits, corresponding to glycogen. Images courtesy of Dr. Clara Salas, Department of Pathology, Hospital Universitario Puerta de Hierro Majadahonda. (Lopes-Sainz 2020) [2].
View Figure 2
Figure 2: Typical findings in PRKAG2 syndrome. (A,B) Hematoxylin-eosin staining displaying hypertrophied myocytes. Magnification 200× and 400×; (C,D) Periodic acid Schiff (PAS) staining positive for glycogen accumulation in cardiomyocyte vacuoles. Magnification 200× and 400×. Yellow arrowheads indicate PAS+ deposits, corresponding to glycogen. Images courtesy of Dr. Clara Salas, Department of Pathology, Hospital Universitario Puerta de Hierro Majadahonda. (Lopes-Sainz 2020) [2].
View Figure 2
Ventricular hypertrophy is the direct result of glycogen deposition and impaired insulin sensitivity. Formed during fetal life, the cardiac annulus fibrosus acts as an electrical barrier isolating the ventricles from the atria. In PS, electrical atrioventricular isolation is compromised due to the disruption in the annulus fibrosus caused by gaps of tissue created by interspersed glycogen-filled myocytes, enabling electrical potentials across, and giving rise to atrioventricular reciprocating arrhythmias and pre-excitation. Cellular autophagy and apoptosis lead to heart failure and advanced conduction disease. SCD may be the result of ventricular fibrillation in young patients with atrial fibrillation and pre-excitation, and older patients suffering complete atrioventricular block [2]. Extracardiac manifestations include systemic arterial hypertension and skeletal myopathy [1].
Proposed “red flags” to help identify PS including shortened PR interval (< 120 ms) with a widened QRS interval (> 110 ms), abnormal initial QRS vector (delta wave), very high voltages, supraventricular arrhythmias, and signs of chronotropic incompetence [1]. Young age of onset, positive family history, myalgias and early-onset hypertension may also be clues [1,6,7]. Other cardiomyopathies associated with WPW, and short PR interval include Danon disease, Anderson-Fabry disease, Pompe disease, Duchenne/Becker muscular dystrophy, MELAS syndrome, Kearns-Sayre syndrome, Leigh syndrome, MERRF, and oncocytic cardiomyopathy [1].
Echocardiography assessment in PS patients usually shows non-specific features including advanced diastolic dysfunction, septal hypertrophy which may be asymmetric, and preservation of LV systolic function and global longitudinal strain [8]. Although cardiac magnetic resonance (CMR) imaging offers tissue characterization patterns that have proven useful in sarcomeric HCM, Anderson-Fabry, and hemochromatosis, no telltale signature findings have been described in PS [9]. CMR has been usual in identifying patients at high risk for SCD based on the extent of late gadolinium enhancement (LGE). The usual threshold for LGE is 15% in HCM patients. Indeed, only histopathology and genetic testing allow clinicians to distinguish PS from its mimickers.
Once the diagnosis is confirmed, close relative genetic cascade testing and proband longitudinal follow up is advised. Suggested guidelines for the management of PS propose to conduct monitoring of disease progression by means of ECG, echocardiogram, and biomarkers on a yearly basis. Prognosis may be assessed via exercise stress testing with oxygen consumption assessment recommended [1].
No significant information is available for primary prevention of SCD by ICDs, however, identification of effort related arrhythmias via exercise stress testing, monitoring arrhythmic burden via device interrogation and/or Holter monitoring, and extent of LGE by CMR may help identify those at increased risk [1]. Ultimately individualized assessment and shared decision techniques should be used when considering implantation of ICD.
Standard treatments for heart failure are recommended while avoiding over diuresis [1]. A few original investigations on animal models have shed light on the role of genetic therapy [10,11], and a recent case report of the serendipitously positive findings with alglucosidase alfa enzyme replacement (rhGAA, Alglucosidase alfa, Genzyme, Boston, MA, USA) offer promising hope [4]. This same drug has been shown to have benefit in type III glycogen storage disease and mucopolysaccharidosis [12,13]. In the research laboratory, the sodium-glucose linked transporter 2 (SGLT2) inhibitor Empagliflozin has been shown to reduce the expression of mRNA of GLUT4 [14], an effect that may be beneficial in PS patients. Whether these and other therapies' potential benefits translate into desirable outcomes remains unknown and should be investigated in clinical trials. Providing referral to physical therapy and rehabilitation specialists is recommended to assess the presence of concomitant skeletal myopathy [1].
Cardiac hypertrophy, conduction disease, and pre-excitation are some of the hallmarks of PS syndrome. Familiarity with these “red flags” is needed to avoid misdiagnosis as its prognosis is poorer than that of sarcomeric HCM. Genetic testing helps distinguish PS from its mimickers. LGE and arrhythmic burden may guide decisions on ICD SCD primary prevention. Clinical trials with SGLT2 inhibitors and alglucosidase alfa replacement are warranted as potential therapeutics for the PS syndrome.
None.
None.
All authors contributed equally to the creation of this case report.