Danon disease is a rare X linked dominant disease that is predominantly affects cardiac muscles; however, skeletal muscles involvement and mental retardation are variable associated features.
We encountered a case of global developmental delay (speech, cognitive and visual) with elevated creatine phosphokinase (CPK) and liver enzymes. He has a strong family history of cardiac disease from the maternal side. Genetic testing showed a hemizygous variant mutation in the lysosome-associated membrane protein-2(LAMP2) gene that was consistent with Danon disease. We find reporting this case should alert cardiologists to screen the at risk family members in order to provide preventive measures.
A 13-years-old boy. He was born at home at 30 weeks of gestation. He stayed in nursery for 3 months. He sat independently at the age of one year, walked at around 3 years and started to run around 4 years of age. Currently, he can walk, and run well. He wears his clothes, button, and unbutton his shirts. He copies letters, scripts and shapes however; he usually does not follow lines. He spoke at the age of 2 years it was single words like mama and dada.
He was barely communicate with his family members at age of 5-6 years; currently he is speaking fluently. He likes to play with sibling and cousins, and he enjoys being with younger children. He enjoys playing football and making new friends. He has a friendly personality. At 11-years of age, he was diagnosed by pediatric neurology to have seizure disorder for which he was started on LevETIRAcetam. Academically, he studies at a regular school at the age of 6-years he was placed at a regular school however; he was soon expelled after few months, and admitted to a special school for children with intellectual disabilities. He repeated the 1st grade three times, 2nd grade twice and 3rd grade 3 times then the father withdrew him from school to another special school where he was able to pass the 6th grade. He has his self-identity and awareness and he knows his age and gender. At 11 years of age, he was diagnosed by pediatric neurology to have seizure disorder for which he was started on LevETIRAcetam.
On examination, his weight was 47 kg (25th-50th centile for age) and his height was 166 cm (50th-75th centile for age). His head circumference was 57 cm (10th-25th centile for his age). He was wearing glasses. He was not dysmorphic. He was having normal Ear, Nose and Eye exam. He was having normal gait and normal power in his upper as well as his lower extremities with normal tone and reflexes.
He had normal heart sounds, normal chest, and abdominal exam. He was having normal circumcised male genitalia with pubertal tanner stage III. He was having normal back and musculoskeletal exam with No gross, bony or soft tissue deformities.
He had normal complete blood count and electrolytes as well as normal renal profile. Liver enzymes were elevated as well as his creatine kinase, his AST was: 486 (0-80 U/L), ALT was 322 (5-45 U/L), and Creatine Kinase was 1783 (30-200 U/L). He had normal Hepatitis serology, GTP. He had normal abdominal Ultrasound (US) and liver Doppler Ultrasound.
His Electrocardiography (ECG) showed essentially normal recording in awake and sleep states. The Cardiac 24 hrs, Holter monitor recorded one episode of second-degree atrioventricular (AV) block Mobitz Type 1 (Wenckebach) otherwise; no other significant arrhythmias were seen.
And his cardiac echogram showed mild concentric left ventricular hypertrophy, The left ventricular posterior wall end systole (LVPWs) is 2.3 cm with Z- score 5.6 and Interventricular septal end diastole (IVSd) is 1.2 cm with Z- score 3.3. Mild tricuspid valve insufficiency with estimated peak gradient of 22 mmHg. Trivial mitral valve insufficiency. No (Left Ventricular Outflow Tract) LVOT/(Right Ventricular Outflow Tract) RVOT obstruction. Un-obstructed aortic arch. Normal left ventricular systolic function.
He was evaluated by genetic service who expected him to have an X-linked disorder based on his family pedigree in which the cardiac disease was following an X-linked inheritance pattern (Figure 1). He has maternal Aunt and uncle who died due to cardiac diseases as well as two maternal aunts who have cardiac diseases but refused to be genetically tested. The tandem MS, plasma amino acids, and urine organic acids were within normal levels. He had normal lactic acid and ammonia. He had normal brain computed tomography (CT) and normal Magnetic resonance imaging (MRI).
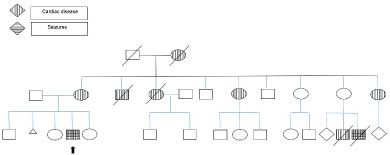 Figure 1: Family Pedigree.
View Figure 1
Figure 1: Family Pedigree.
View Figure 1
The Whole exome sequencing (WES) was requested and found to have a hemizygous variant c.669T>G p. (Tyr223') mutation in the LAMP2 gene (NM_001122606.1): Which is consistent with Danon disease.
Cardiomyopathies are important causes of morbidity and mortality among the pediatric population.
Danon disease is an X linked dominant disease that is caused by mutation in the gene encoding lysosome-associated membrane protein-2 (LAMP2 gene) on chromosome Xq24.
The disorder predominantly affects cardiac muscles; however, skeletal muscles involvements and mental retardation are variable features. It manifests predominantly in young males with a classic triad of cardiomyopathy, skeletal myopathy, and intellectual disability. Females show milder phenotypes with late onset cardiac symptoms with sudden death that usually occurs in their forties [1,2]. Death from cardiac disease is the ultimate cause of demise in many patients if left untreated. The accumulation of glycogen in muscle and lysosomes originally led to the classification of Danon disease as a variant of glycogen storage disease type II with 'normal acid maltase' or alpha-glucosidase. However, Danon disease is not a glycogen storage disease because glycogen is not always increased [3].
In the differential of cardiomyopathy comes the early onset (infantile) glycogen storage disease type II (Pompe disease) which is characterized by hypotonia, hypertrophic cardiomyopathy, hepatomegaly, and respiratory insufficiency typically leading to death within the first year of life without enzyme replacement therapy [4,5]. In contrast, late-onset Pompe disease (juvenile and adult) shows slowly progressive proximal muscle weakness and respiratory difficulties [6,7]. Smaller proportion of patients with late-onset Pompe disease (< 10%) have cardiovascular involvement, including electrophysiological abnormalities and myocardial hypertrophy [8,9].
Another differential for Danon disease is the X linked Excessive Myopathy Autophagy, XEMA which is an X-linked recessive skeletal muscle disorder characterized by childhood onset of progressive muscle weakness and atrophy primarily affecting the proximal muscles. While the onset is usually in childhood, it can range from infancy to adulthood. Many patients lose ambulation and become wheelchair-bound. Other organ systems, including the heart, are clinically unaffected but muscle biopsy shows intracytoplasmic autophagic vacuoles with sarcolemmal features and a multilayered basal membrane [10,11,12].
Daniels MJ, et al. proposed a common patho-physiological basis for the metabolic and structural effects of this descriptive class of heart disorders, and contend that troponin-I may have prognostic value and merits exploration for clinical decision-making including health warning bracelets. Also, they proposed that Rapamycin, an approved immunosuppressant, which influences autophagy, may prove beneficial [13].
Samad F1, et al. reported the aggressive cardiac phenotype of Danon disease that presented with rapid progression to end-stage cardiomyopathy. They found that this progression occurred in both men and women [14].
In Conclusion, Danon Disease is a rare genetic disorder that may not be recognized throughout generations. It causes sudden death that may be correlated to another etiology. We think reporting this case should alert cardiologists to investigate for this rare condition and implement screening for relatives of the affected individuals in order to ensure early counselling and intervention.