The authors hypothesize that thrombosis causes both the complications of atherosclerosis as well as the underlying lesion, the atherosclerotic plaque. Atherosclerotic plaques develop from the organization of mural thrombi. In this process, circulating progenitor cells trapped within the thrombus differentiate into myofibroblasts which synthesize collagen, and endothelial cells which form granulation tissue. Eventually, all or most of the thrombus is replaced by collagen. Thus, conditions and drugs which affect thrombosis also affect the development of atherosclerotic plaques, i.e., atherogenesis. The authors review the process of organization and evidence showing that altered hemodynamics, increased blood viscosity, thrombophilias, coagulopathies, myeloproliferative disorders and drugs which modify the risk of thrombosis also affect atherogenesis. Accelerated atherogenesis is an underappreciated complication of many of these conditions. This paradigm explains how diverse risk factors, many of which have no association with inflammation or dyslipidemia, cause the same lesion. These data have implications for atherothrombosis theory and clinical practice.
Atherothrombosis, Blood viscosity, Atherosclerotic plaque, Hypercoagulability, Bleeding disorder, Mural thrombus, Hemodynamics
The authors hypothesize that atherosclerotic plaques develop from the organization of mural thrombi as originally proposed by the nineteenth century pathologist Carl von Rokitansky. In this paper, the authors review the process of thrombus organization and examine the evidence that flow abnormalities, hematologic abnormalities and pharmaceuticals, all of which modify the risk of thrombosis, also affect atherogenesis (Figure 1). The impact on atherogenesis of some of these factors has not been widely appreciated. Many of these factors foster atherothrombosis by increasing blood viscosity. This paradigm refines the understanding of how risk factors such as blood flow abnormalities and blood properties caused by increased levels of Lp(a) and fibrinogen affect atherothrombosis, and reinforces the fact that splenectomy, thrombophilias and hypofibrinolysis should be recognized as risk factors for atherogenesis.
 Figure 1: Overview of atherogenesis. Many risk factors for accelerated atherogenesis foster mural thrombosis by increasing blood viscosity or directly promoting thrombosis.
Figure 1: Overview of atherogenesis. Many risk factors for accelerated atherogenesis foster mural thrombosis by increasing blood viscosity or directly promoting thrombosis.
Abbreviations: LDL: Low density lipoprotein; HTN: Hypertension; DM: Diabetes mellitus; Hyperfib: Hyperfibrinogenemia; A.M: Antemeridian; Lp(a): Lipoprotein(a); PV: Polycythemia vera; ET: Essential thrombocythemia; Hypofibrin: Hypofibrinolysis; Hypergam: Hypergammaglobulinemia; Erythro def: Erythrocyte deformability; ESA: Erythropoiesis stimulating agents; OC: Oral contraceptive; APS: Antiphospholipid syndrome; Sticky PS: Sticky platelet syndrome; PAI-1: Plasminogen activator inhibitor-1; Thrombo: Thrombophilias.
View Figure 1
The thrombogenic theory of Rokitansky became widely accepted in the mid twentieth century due to the work of Welsh pathologist, John Duguid. When examining serial histologic sections, Duguid observed thrombus in continuity with an atherosclerotic plaque, suggesting the former gives rise to the latter [1]. Thus, thrombosis causes atherothrombotic complications such as myocardial infarction and ischemic stroke, as well as atherogenesis, the formation of the underlying plaque [2,3]. The thrombogenic hypothesis dominated atherogenesis theory until the evolving awareness that hypercholesterolemia is a risk factor for atherothrombosis. This resulted in hypercholesterolemia assuming the central role in atherogenesis theory despite the fact that diabetes mellitus, hypertension, cigarette smoking, and physical inactivity are stronger risk factors for coronary heart disease [4]. We have recently reviewed the deficiencies in mainstream atherothrombosis theory [3,5].
The thrombogenic theory emphasizes abnormal blood flow instead of putative inflammatory effects of cholesterol, which is a component of all animal cell membranes. Like occlusive thrombi in deep leg veins, mural thrombi develop from Virchows's triad: Abnormalities in the arterial wall, disturbed patterns of blood flow, and abnormal composition of blood. Atherothrombosis can be viewed as beginning with the loss of aortic elasticity. This occurs with aging and accelerates with hypertension, diabetes, male gender, and menopause. Aortic stiffness is a strong independent risk factor for cardiovascular events [6], which is not explained or even addressed appropriately by mainstream theory. The protein elastin contributes significantly to aortic elasticity and is encoded by ELN. ELN genotype has a significant effect on carotid intima-medial thickness [7].
Aortic elasticity dampens peak aortic blood velocity, producing slower flow over a larger fraction of systole. Loss of aortic elasticity increases peak velocity [8]. This increases Reynolds number, which estimates the probability of developing flow separation with changing arterial geometry. In areas of flow separation, a portion of the blood flow separates from the bulk blood flow and its velocity greatly decreases [2]. Thus, high velocity flow is in close proximity to low velocity flow (Figure 2 and Figure 3) [9]. A familiar example of this phenomenon is the development of eddy currents when fast flowing water encounters a rock.
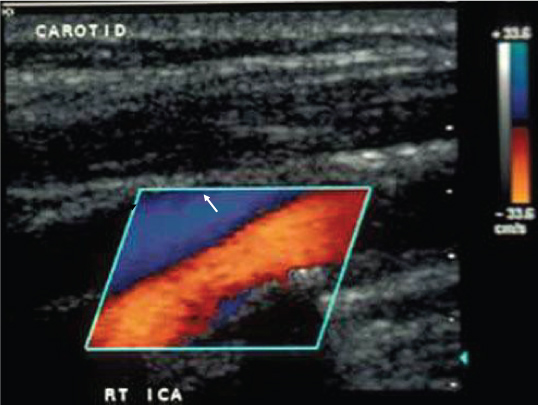 Figure 2: Altered hemodynamics caused by an atherosclerotic plaque. Color ultrasound of the right internal carotid artery and jugular vein. The direction of flow and velocity can be determined from the scale in the upper right. Flow in the carotid (red and yellow) is higher velocity and in the opposite direction of flow in the jugular vein. Flow in the carotid accelerates as it encounters a stenosis (white arrow). The stenosis creates an extreme hemodynamic environment which creates an area of flow separation distally (black arrow). Flow in the area of separation is slow and retrograde to the bulk flow. The area of flow separation is prone to thrombosis because platelets are activated by shear in the high velocity flow and tend to aggregate in the area of separation. Modified from reference [2]. Courtesy of Robert S. Perret, M.D.
View Figure 2
Figure 2: Altered hemodynamics caused by an atherosclerotic plaque. Color ultrasound of the right internal carotid artery and jugular vein. The direction of flow and velocity can be determined from the scale in the upper right. Flow in the carotid (red and yellow) is higher velocity and in the opposite direction of flow in the jugular vein. Flow in the carotid accelerates as it encounters a stenosis (white arrow). The stenosis creates an extreme hemodynamic environment which creates an area of flow separation distally (black arrow). Flow in the area of separation is slow and retrograde to the bulk flow. The area of flow separation is prone to thrombosis because platelets are activated by shear in the high velocity flow and tend to aggregate in the area of separation. Modified from reference [2]. Courtesy of Robert S. Perret, M.D.
View Figure 2
 Figure 3: Flow separation caused by an airfoil in a wind tunnel. The tendency to develop flow separation (white arrows) is predicted by Reynolds number, which increases with the velocity of flow. Higher velocity blood flow in systole is created by aortic stiffening, which accelerates in hypertension, diabetes, males and menopause. It is a natural consequence of aging. Courtesy of Deutsches Zentrum für Luft- und Raumfahrt.
View Figure 3
Figure 3: Flow separation caused by an airfoil in a wind tunnel. The tendency to develop flow separation (white arrows) is predicted by Reynolds number, which increases with the velocity of flow. Higher velocity blood flow in systole is created by aortic stiffening, which accelerates in hypertension, diabetes, males and menopause. It is a natural consequence of aging. Courtesy of Deutsches Zentrum für Luft- und Raumfahrt.
View Figure 3
Atherosclerotic plaques develop in areas of low shear or sluggish blood flow created by this flow separation [10]. Areas of slow blood flow are predisposed to thrombosis because of decreased shear-mediated endothelial synthesis of anti-thrombotic molecules like prostacyclin and nitric oxide, causing the phenomenon recognized as "endothelial dysfunction". Activated coagulation factors accumulate and influx of anticoagulant factors is decreased in these areas of low shear. The high velocity blood flow against the opposite arterial wall protects it from thrombosis, limiting growth of the thrombus to one arterial wall. Therefore, arterial thrombi tend to be mural, not occlusive. Quite the difference is found in blood flow in veins. Blood flow in veins is too slow to create flow separation so that thrombi become occlusive. Thus, atherosclerotic plaques occur only in arteries.
Blood viscosity must also be considered in this discussion. Increased blood viscosity may be the most common abnormality in the composition of blood which predisposes to thrombosis. The commonly recognized risk factors for atherothrombosis such as hypercholesterolemia, hypertension, diabetes mellitus, cigarette smoking, obesity, hyperfibrinogenemia, aging, and male gender, are associated with increased blood viscosity [11]. Increased blood viscosity causes the development of larger areas of even slower blood flow. This is a manifestation of the "non-Newtonian" nature of blood viscosity, i.e. the viscosity of blood is not constant but increases sharply when shear rate decreases (Figure 4). When measured near stasis (flow velocity of 0.1 mm/sec), blood viscosity in patients with thrombosis is five to ten times higher than in healthy subjects. In healthy subjects, blood viscosity decreases from approximately 1,000 centipoise when near stasis to 5 centipoise at flow velocities of 10 to 100 cm/sec [12].
 Figure 4: The relationship of blood viscosity to shear rate. Blood viscosity is maximal at low shear rates (left side of diagram) as a consequence of reversible erythrocyte aggregation. Blood viscosity decreases as rate of shear increases because of disaggregation and erythrocyte deformability. Erythrocytes assume an ellipsoidal shape to minimize resistance to flow at high shear rates. Factors which influence blood viscosity are shown in relation to the shear domain in which they are active.
Figure 4: The relationship of blood viscosity to shear rate. Blood viscosity is maximal at low shear rates (left side of diagram) as a consequence of reversible erythrocyte aggregation. Blood viscosity decreases as rate of shear increases because of disaggregation and erythrocyte deformability. Erythrocytes assume an ellipsoidal shape to minimize resistance to flow at high shear rates. Factors which influence blood viscosity are shown in relation to the shear domain in which they are active.
Abbreviations: WPV: Whole blood viscosity; mP: Millipoise; Fib: Fibrinogen.
Courtesy of www.BloodFlowOnline.com.
View Figure 4
Increased blood viscosity at low shear rates is due to reversible erythrocyte aggregation. Molecules or particles large enough to span the minimum intercellular distance between two erythrocytes, such as fibrinogen, low density lipoprotein (LDL), or lipoprotein(a) (Lp(a)), can simultaneously bind to two erythrocytes and foster aggregation [13]. Because it is too small to simultaneously bind to two erythrocytes, high density lipoprotein (HDL) decreases blood viscosity by competing with LDL for erythrocyte binding. The non-Newtonian behavior of blood raises the possibility of a vicious cycle: A decrease in blood velocity allows an increment of erythrocyte aggregation which will increase blood viscosity, which will further decrease flow allowing further aggregation, higher viscosity, slower flow, more erythrocyte aggregation, etc. This possibility of this scenario is increased in blood with high concentrations of LDL, Lp(a) or fibrinogen.
The hemodynamics described above also cause the phenomenon known as the "vulnerable plaque". An atherosclerotic plaques causes an increase in the velocity of blood flowing over it, which will cause shear-mediated platelet activation and flow separation distally. Nascent atherosclerotic plaques which form in this hemodynamic extreme may develop superimposed thrombosis before fibrous remodeling is complete, resulting in a symptomatic plaque with a thin fibrous cap, a large lipid core (vide infra), prominent neovascularization, and a lesser degree of stenosis. These are the characteristics of a vulnerable plaque [2,3].
Increased blood viscosity can also cause elevations of C-reactive protein (CRP), which have been accepted as evidence that atherothrombosis is an inflammatory disease. Because perfusion is inversely proportional to vascular resistance, increased blood viscosity, which increase vascular resistance, reduces perfusion of skeletal muscle. This will reduce glucose uptake by myocytes, reduce cytoplasmic glucose in myocytes, and prolong postprandial hyperglycemia. As a homeostatic response to decreased cytoplasmic glucose, myocytes synthesize the myokine interleukin-6, which initiates a homeostatic cascade that increases plasma levels of insulin, tumor necrosis factor-alpha and CRP [3].
Organization is the process by which thrombi, exudates, and hematomas are remodeled into fibrous tissue. In this process, circulating progenitor cells of bone marrow origin are trapped within thrombi and differentiate into myofibroblasts which synthesize collagen and endothelial cells which form granulation tissue [14]. Cells of bone marrow origin have been identified in coronary lesions in recipients of sex-mismatched bone marrow [15] and cardiac allografts [16]. At a minimum, the organization of mural thrombi is acknowledged to add to the volume of an atherosclerotic plaque [17].
The progression of atherosclerotic plaques on serial coronary angiography is consistent with thrombogenic theory. Lesion progression is episodic, not linear, as would be expected if atherogenesis were a chronic, low-grade inflammatory process which begins in infancy with the development of fatty streaks. High-grade stenosis frequently develop on normal arterial segments, and most pre-existing lesions remain stable [18]. These findings also show that the distinction between primary and secondary prevention is largely arbitrary. The process which causes the development of a new high grade stenosis in a patient with a previous myocardial infarction is the same which caused the lesion that initially brought the patient to medical attention. Further, the likelihood of developing a new atherosclerotic plaque is increased by progressive aortic stiffening, increased blood viscosity with aging, and the adverse hemodynamic milieu distal to the initial atherosclerotic plaque. Thus, "secondary prevention" is more difficult to achieve than primary prevention. The prevention of additional adverse cardiac events in patients treated with the oral anticoagulant rivaroxaban after an initial adverse coronary event is evidence of the role of thrombosis in atherogenesis [19,20].
A growing body of literature has examined the role of platelets in the initiation of atherothrombosis, supporting an origin of atherosclerotic plaques from mural thrombi [21-24].
Completely organized mural thrombi and atherosclerotic plaques are morphologically indistinguishable [25]. The origin of atherosclerotic plaques from mural thrombi is demonstrated by plaques in synthetic arteriovenous grafts. These plaques are common, being present in 59 of 78 grafts in one study [26]. In these grafts, very high velocity blood flow results in platelet activation, flow separation, and a predisposition for atherothrombosis.
Like plaques in native arteries, atherosclerotic plaques in synthetic grafts are eccentric collagenous lesions which contain myofibroblasts, neovascularization, foam cells, dystrophic calcifications, and eventually develop superimposed thrombi. Immunoperoxidase staining for smooth muscle actin reveals both morphologic types of smooth muscle cells seen in atherosclerotic plaques in native arteries: Elongated pancake-shaped cells and plumper cells with intense peripheral staining (Figure 5A and Figure 5B). Several authors have described atherosclerotic plaques in synthetic vascular grafts [27-30]. For example, Carson, et al. noted the similarity of a lesion which partially occluded an entire polytetrafluoroethylene graft to the plaques in the patient's native arteries [27]. Experimental fibrin emboli in rabbits become covered by endothelium within four days and incorporate collagen after six days. Granulation tissue completely replaces fibrin in a week, and after a month only fibrous tissue remains [31]. Duguid demonstrated that fibrin deposits are common on intact intima. He found fibrin deposits on the intima of 19 of 50 consecutive aortas from subjects aged 3 to 73 [32]. His work suggests that physical damage to the endothelium is not necessary for thrombosis. Subtler abnormalities such as endothelial cell loss due to low shear [10] may be sufficient.
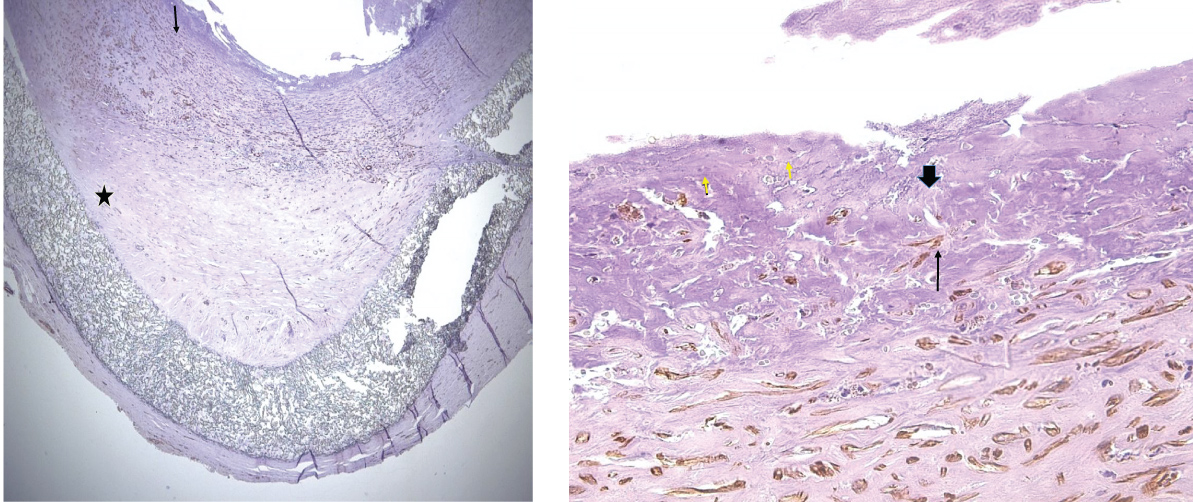 Figure 5: Dacron arteriovenous graft stained for smooth muscle actin. Atherosclerotic plaque from a dacron arteriovenous graft stained for smooth muscle actin (SMA). Positive cells are brown. A) Low power image. Arrow indicates a thin layer of fibrin on the luminal surface of the plaque. The graft is indicated by a star. Original magnification: 20×. B) High power photomicrograph of the fibrin layer. Short arrows indicate SMA positive cells in the fibrin layer. Long and block arrows indicate positive cells in the fibrous plaque. Block arrow points to a "pancake" cell. Long arrow points to a plumper cell. Original magnification: 400×. Immunoperoxidase staining protocol for smooth muscle actin per ref. [26].
View Figure 5
Figure 5: Dacron arteriovenous graft stained for smooth muscle actin. Atherosclerotic plaque from a dacron arteriovenous graft stained for smooth muscle actin (SMA). Positive cells are brown. A) Low power image. Arrow indicates a thin layer of fibrin on the luminal surface of the plaque. The graft is indicated by a star. Original magnification: 20×. B) High power photomicrograph of the fibrin layer. Short arrows indicate SMA positive cells in the fibrin layer. Long and block arrows indicate positive cells in the fibrous plaque. Block arrow points to a "pancake" cell. Long arrow points to a plumper cell. Original magnification: 400×. Immunoperoxidase staining protocol for smooth muscle actin per ref. [26].
View Figure 5
Obviously, synthetic grafts have a limited ability to respond to injury. They do not develop fatty streaks or have a tunica media as a source for putative smooth muscle migration into an atherosclerotic plaque, demonstrating that a mural thrombus, circulating progenitor cells, and persistence of the thrombus for several days are sufficient for atherogenesis. In contrast, the mainstream response to injury hypothesis holds that fatty streaks are the precursor to atherosclerotic plaques. Received wisdom holds that atherogenesis begins with mass transport of LDL across the endothelium and insudation in the intima, followed by oxidative modification, foam cell formation, endothelial dysfunction, smooth muscle migration from the tunica media, fibrosis, and superimposed thrombosis.
This pathway is problematic for several reasons. Insudation of LDL cannot explain the localization of atherosclerotic plaques to areas of low shear in the arterial tree. The purpose of the endothelium is to prevent thrombosis, not serve as an impermeable barrier. Although it is not widely appreciated, normal endothelium is semi-permeable [33]. All plasma macromolecules, including LDL, are present throughout the arterial intima in concentrations directly proportional to their molecular weight and plasma concentrations. Molecules which bind and retain LDL, such as collagen, fibronectin, and proteoglycans, are present throughout the arterial intima as components of diffuse intimal thickening, a universal change in the arteries of adults [17]. Mass transport across the venous endothelium is equal to or greater than across the arterial endothelium [34], yet veins do not develop atherothrombosis. The presence of LDL and all other components of plasma in the subendothelial space is physiologic, not pathologic, and a mechanism to remove them has evolved (vide infra).
Second, fatty streaks routinely resolve without sequelae [35]. Further, oxidized LDL is present in both human arteries and veins, and its presence does not correlate with the pathologic effects suggested by in vitro data [36]. Finally, the lipid content of a fatty streak is different from that in the core of an atherosclerotic plaque. Fatty streaks have a higher content of cholesteryl ester than free cholesterol, a low sphingomyelin to lecithin ratio, and a high oleate to linoleate ratio [37]. Enrichment of oleate in fatty streaks is due to the increased binding of LDL particles enriched in cholesteryl oleate to the proteoglycan biglycan [38]. These lipid ratio are reversed in the core of an atherosclerotic plaque. The differences in the lipid content of fatty streaks and the core of an atherosclerotic plaque were identified in the 1970's and have been confirmed [37]. However, this knowledge has not been incorporated into the response to injury theory.
Like the lipid core of an atherosclerotic plaque, the erythrocyte cell membrane contains more unesterified than esterified cholesterol and a high content of sphingomyelin [39]. The lipid content of the core of an atherosclerotic plaque can be explained by an origin from the erythrocyte cell membrane and the absence of proteoglycans to bind LDL enriched in oleate. Further evidence of an origin from the erythrocytes are the presence of hemosiderin and glycophorin A in the lipid core [40]. These observations support the origin of an atherosclerotic plaque from organized mural thrombus, not a fatty streak. The lipid core of an atherosclerotic plaque originates from degenerated thrombus which has not undergone complete fibrous remodeling, or intraplaque hemorrhage.
Advances in cell biology since Russell Ross proposed the response to injury theory of atherogenesis in the 1970's have elucidated the physiologic process by which LDL and cholesterol are removed from the intima. Following movement across the endothelium, plasma components in the subendothelial space are taken up by dendritic cells via macropinocytosis, a process which does not require a receptor or oxidative modification. Dendritic cells are a normal component of the arterial intima, and can pinocytose 100 times their volume per hour in vitro. This great capacity for macropinocytosis is necessary for dendritic cells to fulfill their role as immunologic sentinels and antigen processors. Dendritic cells do not possess enzymes capable of catabolizing cholesterol. Instead, they package cholesterol into membrane-bound vesicles called exosomes which are secreted into the extracellular space and circulate until cleared by reticuloendothelial cells of the liver. In areas where the endothelium is compromised by low shear, increased movement of lipoproteins across the endothelium causes dendritic cells to accumulate cholesterol faster than it can be eliminated and they assume the appearance of foam cells. Elimination of cholesterol from the intima is described in greater detail in reference [3]. Inflammation, monocyte chemoattraction, oxidative stress, and reverse transport of cholesterol play no role in the formation or resolution of fatty streaks. Significantly, monocyte transmigration in association with fatty streaks, oxidative stress, smooth muscle migration, and reverse transport of cholesterol from fatty streaks have not been demonstrated in vivo in humans. Oxidative stress and reverse transport of cholesterol are ad hoc modifications of the response to injury theory added to explain the associations of LDL and HDL with their risk of atherothrombosis. Some suggest that ad hoc modifications to a theory decrease its prestige [5].
Results from mouse knockout models and the Watanabe heritable hyperlipidemic rabbit have been interpreted as supporting the response to injury hypothesis. The gross arterial lesions in these models viewed en face can be visualized by staining with Sudan IV, which stains lipids (for example, see ref. [41]). Human atherosclerotic plaques are not consistently sudanophilic because the lipid core, if present, is covered by a fibrous cap. Instead, these appear grossly as raised white to off-white lesions. Sudanophilia is not required for their identification [42]. In the experience of one author (GDS) who has examined hundreds of aortas and coronary arteries from the Pathobiological Determinants of Atherosclerosis in Youth (PDAY) archive, sudanophilia practically never overlies or obscures an atherosclerotic plaque. Histopathologically, the arterial lesions in the mouse and rabbit models are also seen to be fatty streaks, not atherosclerotic plaques. Schwartz, et al. has discussed the histopathologic differences between human atherosclerotic plaques and its supposed murine equivalent [43]. Proponents of mouse models argue that these histopathologic differences are unimportant [44,45]. Further, hemodynamic variables such as vessel diameter, wall shear stress, and heart rate markedly differ in mice and humans. The smooth muscle cells in mouse lesions are derived from the vessel wall, whereas at least some smooth muscle cells in human lesions are derived from the bone marrow [45]. Thus, the pathogenesis of murine "fibrous plaques" is different from the pathogenesis of human atherosclerotic plaques. Therefore, extrapolation from murine models to human atherothrombosis is inappropriate.
The association of single nucleotide polymorphisms (SNP) with atherothrombosis has also been argued to support the response to injury hypothesis. For example, polymorphisms in SCARB1 are associated with variations in the plasma lipid profile [46]. This type of data shows that an abnormality associated with a disease is causally related to that disease, and not a manifestation of the disease. Thus, this type of data rules out reverse causation. SNPs show that abnormal plasma lipids are one cause of atherothrombosis, but not the mechanism by which they cause the disease. Polymorphisms of the gene for endothelial nitric oxide synthase which decrease production of nitric oxide, a molecule with anti-thrombotic activity, support the thrombogenic theory. The polymorphisms G894T and T786C are significantly more frequent in subjects with a first myocardial infarction before age 35 than age-and sex-matched controls [47].
Supporting the assertion that thrombosis is the underlying abnormality in atherogenesis, Prandoni, et al. found an associated between spontaneous deep venous thrombosis (DVT) and asymptomatic carotid atherosclerosis diagnosed ultrasonographically [48]. At least one carotid atherosclerotic plaque was found in 72 of 153 subjects with spontaneous DVT (47.1%), but only 48 of 150 controls (32.0%). The odds ratio for subjects with spontaneous DVT of having a carotid plaque as compared to controls was 2.3 (95% confidence interval (CI): 1.4 to 3.7). In a multivariate analysis which accounted for age, sex, smoking status, obesity, hypertension, hyperlipidemia, and diabetes, the strength of the association did not change. Thrombophilias were found in 25 of 68 subjects who underwent screening (36.8%). Meta-analyses in 2008 [49] and 2016 [50] confirmed the association between the risk factors for venous thromboembolism (VTE) and atherosclerosis.
The major risk factors for atherothrombosis, i.e., age, hypertension, obesity, diabetes mellitus, metabolic syndrome [51,52], and hypercholesterolemia [53] are also risk factors for arterial and venous thrombosis, including DVT. These risk factors are all associated with elevated blood viscosity [3,11]. Elevated blood viscosity was shown to be an etiological factor for the development of DVT in a study in which blood viscosity was measured pre-operatively in 52 general and gynecologic surgery patients. Blood viscosity in the eleven patients who developed DVT was 21% higher than in those who did not [54]. Historically, the precise cause of DVT in 30% of patients is unexplained by traditional risk factors for this condition [48]. Unrecognized blood hyperviscosity may explain many of these cases.
Elevated blood viscosity also plays a role in arterial thrombosis. Because the high rates of venous and arterial thrombosis in systemic lupus erythematosus (SLE) are not explained by traditional risk factors, Booth and colleagues measured blood viscosity in 60 subjects with SLE, 30 of whom had a thrombotic event. Seventeen events were arterial. In these, blood viscosity was higher than in the other SLE patients (p = 0.014) and controls (p = 0.022). There was no difference in blood viscosity between those who received anticoagulation with warfarin or heparin versus those who didn't. Presence or absence of antiphospholipid antibodies and prednisone dose had no impact on blood viscosity [55].
Elevated blood viscosity is a risk factor for atherothrombosis as well. Lowe and coworkers measured blood viscosity, hematocrit, fibrinogen and plasma viscosity in 70 men prior to undergoing coronary angiography. Their coronary angiograms revealed 26 men had significant stenoses in two or three coronary arteries, and 24 males had a stenosis in one artery or no stenosis. There were 20 healthy controls. Blood viscosity, hematocrit, and fibrinogen concentration were significantly higher in those with stenoses in two or three arteries than the other groups [56].
In addition to creating larger areas of slower blood flow, elevated blood viscosity may be prothrombotic and atherogenic by promoting shear-mediated platelet activation. Because blood viscosity is the ratio of shear stress/shear rate, elevated blood viscosity increases shear stress at all shear rates. The impact of LDL on microthrombus formation was demonstrated by Lacoste and co-workers. They perfused unanticoagulated human venous blood over denuded porcine aortic media at high shear rates and then quantified mural microthrombi microscopically. The number of microthrombi was increased in those with hypercholesterolemia, and decreased to control levels after two to three months of therapy with the HMG-CoA reductase inhibitor, pravastatin [57]. Because pravastatin therapy decreases blood viscosity [58], the greater number of microthrombi prior to statin therapy may have been caused by increased shear-mediated platelet activation due to elevated blood viscosity.
The spleen removes older, stiff erythrocytes. Splenectomy results in decreased erythrocyte deformability, increased blood viscosity, and thrombocytosis [59], causing a thrombophilia involving both the venous and arterial circulations [60]. In World War II servicemen who underwent splenectomy due to trauma, the only causes of death which were increased were coronary artery disease and pneumonia [61]. Because combat soldiers are commonly young, most presumably underwent splenectomy before atherogenesis was advanced.
Splenic immune function has been postulated to promote atherogenesis [62]. The excess deaths due to myocardial infarction after splenectomy argues against this. Splenectomy for hereditary spherocytosis is associated with an increased risk of thromboembolic events [63]. Compared to hereditary spherocytosis patients who did not undergo splenectomy, the hazard ratio for arterial thrombosis post-splenectomy is 7.2 (95% CI: 2.8-7.2). The hazard ratio for venous thrombosis is 3.3 (95% CI: 1.1-9.8). The thrombophilia and increased atherogenesis associated with splenectomy may be due to elevated blood viscosity.
Fibrinogen is a risk factor for venous [64] and arterial [65] thrombosis. Fibrinogen is also a major independent risk factor for atherothrombosis [66]. Even a modest (10%) increase in fibrinogen increases the odds of subsequently developing coronary heart disease [67]. Levenson, et al. examined the association between fibrinogen concentrations and atherosclerotic plaques in the abdominal aorta, carotid and/or femoral arteries in asymptomatic males aged 40 to 60 with at least one conventional risk factor for atherothrombosis. Multivariate analysis showed an independent association between fibrinogen and the presence and extent of atherosclerotic plaques. Plaque prevalence was significantly associated with increasing tertile of fibrinogen levels [68].
Besides increasing plasma viscosity, fibrinogen impacts blood viscosity by augmenting erythrocyte aggregation. This is the basis for the erythrocyte sedimentation rate (ESR), an assay commonly used to measure the severity of clinical inflammation. Because both are affected by erythrocyte aggregation, the ESR closely correlates with low shear blood viscosity. In a meta-analysis, an elevated ESR increased the relative risk of coronary heart disease by 1.33 (95% CI: 1.15 to 1.54) [69]. Thrombophilia and increased atherogenesis associated with hyperfibrinogenemia is due to elevated blood viscosity.
Variations in plasma fibrinogen concentrations and hematocrit contribute to the circadian variation in blood viscosity Plasma protein concentrations and hematocrit are highest on arising, between 0745 and 0800 hrs. Because blood viscosity increases exponentially at lower shear rates, mean blood viscosity at the lowest shear rate can vary between 62 and 136% over 24 hours. Higher blood viscosity may explain the increased incidence of acute coronary syndromes and stroke in the morning hours. It may also be the reason why the efficacy of thrombolytics varies during the day [70].
Fibrinogen, like C-reactive protein (CRP), is an acute phase reactant, and plasma levels increase with inflammation. However, Kamphuisen and colleagues suggest that the elevated concentrations of fibrinogen in venous thrombosis patients are not due to the acute phase reaction. Those investigators found that elevations of fibrinogen > 14.7 μmol/L increased the risk of venous thrombosis four-fold. Although CRP was also elevated in thrombosis patients, the investigators found that controlling for the effect of CRP did not change the increased risk associated with elevated levels of fibrinogen. This suggests that fibrinogen elevations in venous thrombosis patients are independent of increases in CRP [71]. It is estimated that 51% of the variance in fibrinogen concentrations is due to genetic heritability [72], which undoubtedly contributes to the 30-60% estimated heritability of atherosclerosis [73].
The success of the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS) has been interpreted as confirmation of the inflammatory basis of atherothrombosis. This study involved patients at increased risk for a second myocardial infarction or other vascular event because of persistent mild elevations of CRP, ≥ 19 nmol/L. Subjects were randomized to either canakinumab, a monoclonal antibody which neutralizes interleukin-1β, or placebo. Subjects who received 150 mg of canakinumab subcutaneously every three months had a significant decrease in the primary endpoints, non-fatal myocardial infarction, non-fatal stroke, or cardiovascular death. After a median follow-up of 3.7 years, 4.50 events occurred per 100 person-years in the placebo group and 3.86 events per 100 person-years in treated subjects, a 14% decrease (hazard ratio: 0.85 (95% CI: 0.74 to 0.98; p = 0.021). CRP decreased 37% [74]. In an earlier Phase II trial, subcutaneous injection of 150 mg of canakinumab monthly decreased fibrinogen levels from 9.7 μmol/L to 7.6 μmol/L, both normal values, after four months [75]. This 22% decrease in fibrinogen levels will decrease blood viscosity and should decrease the risk of myocardial infarction. Thus, the decrease in adverse events with canakinumab therapy may be due to hemorheological improvement, not decreased inflammation. Fibrinogen levels were not reported in the Phase III study. A more valid test of the inflammatory basis of atherothrombosis may be the Cardiovascular Inflammation Reduction Trial (CIRT), which will test the ability of low-dose methotrexate to decrease reinfarction. Low-dose methotrexate reportedly does not decrease fibrinogen levels [76]. The decrease in CRP in CANTOS could also be due to decreased blood viscosity, as noted above.
Lp(a) is an LDL-like particle which contains an additional apolipoprotein that has an amino acid sequence homology to plasminogen. It is a risk factor for arterial [77] and venous thrombosis [78]. In a study of premature cardiovascular disease, Milgrom, et al. found an increased prevalence of elevated Lp(a) in cases compared to controls (vide infra) [79]. A 2010 review concluded that elevated Lp(a) was a cause of premature atherosclerosis [80]. We have recently argued that Lp(a) causes disease by increasing blood viscosity [81]. Supporting this, Bohl, et al. performed a single session of lipoprotein apheresis, which removes both LDL and Lp(a), in ten subjects with Lp(a) levels > 60 mg/dL. Twenty-four hours post-procedure, the concentrations of several rheologically-active particles and molecules, including LDL, Lp(a), HDL, and fibrinogen were reduced. Plasma viscosity was reduced by 10.7% and ejection fraction increased [82]. Ejection fraction improved because reduced blood viscosity decreases cardiac afterload as dictated by Poiseuille's law (Q = ((∆P × ∏ × r4)/8l) × 1/η in which η = viscosity). In one study, 20% decrease in blood viscosity resulted in a 40% decrease in brain natriuretic peptide (BNP), probably reflecting the non-linear relationship between blood viscosity and shear rate [83]. Bosch, et al. found that a single session of lipoprotein apheresis reduced LDL, Lp(a), and very-low density lipoprotein levels, which was associated with a 10% decrease of plasma viscosity and a 42% decrease in erythrocyte aggregation [84], both of which decrease blood viscosity.
Kamstrup and colleagues attempted to distinguish between the prothrombotic and atherogenic activity of Lp(a) using a Mendelian randomization approach. They reported that only Lp(a) levels > 95th percentile and number of kringle IV repeats < 6th percentile were associated with an increased risk of venous thrombosis. In contrast, tertiles of both Lp(a) concentration and number of kringle IV repeats correlated with coronary, carotid, and femoral atherosclerosis [85]. The limitations of this study were discussed in the accompanying commentary [86].
In 2017, Milgrom, et al. reported the prevalence of thrombophilias in 153 patients aged ≤ 45 with premature cardiovascular disease (CVD). Within this cohort, 79% of patients suffered a myocardial infarction, 46% received a stent, and 35% underwent bypass grafting. The investigators compared the prevalence of thrombophilia in these patients with 110 healthy controls and 110 patients with venous thromboembolism (VTE) without cardiovascular disease. Elevated levels of factor VIII were present in 22% of premature CVD cases vs. only 7% in controls (p = 0.007). Elevated anticardiolipin IgM antibody levels were present in 9% of cases vs. only 2% of controls (p = 0.024), elevated Lp(a) levels were present in 30% of cases vs. 19% of controls (p = 0.038), and lupus anticoagulant was present in 11% of premature CVD cases vs. 2% of the controls (p = 0.035). CVD cases were similar to VTE patients except for a decrease in free protein S in 28% of VTE patients and 11% of the CVD cases (p = 0.03) and a lower frequency of Lp(a) elevation in VTE cases, 17% (p = 0.032). These data demonstrate an increased prevalence of thrombophilia in patients with premature CVD, consistent with a role for thrombosis in atherogenesis. The data also demonstrate significant overlap between the risk factors for VTE and premature CVD, further supporting a role for thrombosis in the etiology of atherogenesis [79].
Vig and coworkers screened 150 patients with symptomatic PVD for markers of thrombophilia. Thrombophilia was present in 41 (27.3%) of these patients. Protein S deficiency was found in 17 (11.3%), factor V Leiden in 10 (6.7%), protein C deficiency, lupus anticoagulant or a prothrombin mutation in 6, and antithrombin deficiency in one. Interpretation of these data is limited because the prevalence of thrombophila was not determined in a control population [87]. Glueck, et al. reviewed the prevalence of thrombophilias and hypofibrinolysis in subjects aged ≤ 45 years of age with atherothrombotic disease [88]. They included patients with angina (41 of 230 events) and claudication (9 of 230 events). Forty-four male and 76 female healthy adults were controls. In males, Factor V Leiden mutations were present in 10% versus 0% in controls, Factor VIII was elevated (> 150%) in 27% versus 2% in controls, Factor XI was elevated (> 150%) in 16% versus 0% in controls, and plasminogen activator inhibitor activity was elevated (> 21.1 U/ml) in 24% versus 7% of controls. All differences were significant, p < 0.02 or less. In women, protein C was decreased (< 73%) in 15% versus 0% of controls, free protein S (< 66%) in 19% of cases versus 3% of controls, Factor XI was high (> 150%) in 11% versus 1% of controls (p = 0.057), and the lupus anticoagulant was present in 28% of cases versus 4% of controls. Except for Factor XI, p vales were 0.014 or less. Besides supporting the role of mural thrombosis in atherogenesis, these data suggest that the prevalence of thrombophilias in asymptomatic people ≤ 45 years is low.
Three autoantibodies are included in the antiphospholipid syndrome (APS): Anticardiolipin, lupus anticoagulant, and anti β2-glycoprotein I. These cause arterial and venous thrombosis by inducing endothelial cell dysfunction [89]. APS is also associated with premature atherosclerosis. In a study of 62 patients aged < 50 years who underwent operations for PVD, the prevalence of anticardiolipin (14.5%) and anti-endothelial antibodies (12.9%) was higher than in controls (p < 0.05). Conventional risk factors were more common in APS patients than controls. However, the prevalence of hyperlipidemia and dyslipidemia was lower in patients with APS [90]. In another study, antiphospholipid antibodies were present in 83 of 262 patients (32%) who underwent bypass grafting for PVD [91]. Progression of PVD is also faster in subjects with antiphospholipid antibodies [92], probably because of the organization of thrombus superimposed on a pre-existing atherosclerotic plaque, which is accepted to contribute to plaque volume [20].
Sticky platelet syndrome is a rare, autosomal dominant disorder which causes platelet hyperaggregability. Clinical symptoms include angina pectoris, myocardial infarction, and venous thrombosis. Arterial thromboses develop without evidence of endothelial injury. Patients usually seek treatment for transient or permanent arterial occlusion [93]. The syndrome was initially described in patients with coronary artery disease, transient ischemic attacks/strokes, or ischemic optic neuropathy without any known risk factors [94].
Nephrotic syndrome is protein-losing nephropathy which increases the risk for arterial and venous thrombosis [95] and premature atherosclerosis. Patients are thrombophilic because of the loss of antithrombin III (AT III) and plasminogen. AT III inhibits coagulation by binding to and inactivating thrombin. Plasminogen is the proenzyme of plasmin, the enzyme which breaks down fibrin and lyses thrombi. LDL and alpha 2-macroglobulin concentrations are increased, which also contributes to hypercoagulability. Alpha 2-macroglobulin inhibits plasmin, worsening hypofibrinolysis. Curry Jr and Roberts examined the coronary arteries of 20 patients with nephrosis, 15 to 35 years of age, at autopsy. The lumen of one or more of the four major coronary arteries was found to be narrowed > 75% in 8 of 20 nephrotic patients and 0 of 14 controls. In nephrotic patients, the lumen of 88 cm of coronary artery (30% of total length examined) was > 50% narrowed, whereas only 5.5 cm of 288 cm (2%) of coronary artery in controls was narrowed to this degree [96]. Kallen, et al. reported the case of a nephrotic five-year-old with severe atherosclerosis of both coronary arteries [97]. Hypoalbuminemia, another manifestation of the nephrotic syndrome, increases blood viscosity and the risk of thrombosis by altering the lipid composition of the erythrocyte cell membrane, which decreases erythrocyte deformability. Nephrosis is also associated with hyperfibrinogenemia and hypertriglyceridemia, both of which increase blood viscosity [98].
Ideally, a thrombus is dissolved by the fibrinolytic system before it organizes. Plasminogen activator inhibitor-1 (PAI-1) is the principal inhibitor of enzymes which activate plasmin, preventing fibrinolysis. Thus, increased PAI-1 activity promotes remodeling of a mural thrombus into an atherosclerotic plaque. Elevated concentrations of PAI-1 are present in the metabolic syndrome.
Levy, et al. studied 51 consecutive patients with PVD before age 45. Elevated levels of PAI-1 were found in 59%, tissue plasminogen activator deficiency (following stimulation of synthesis by 20 minutes of vascular occlusion) in 45%, and elevated Lp(a) in 86% of patients [99]. The prevalence of these factors was not determined in controls. Tsitouris, et al. studied five patients with acute myocardial infarction and 9 patients with angina and ECG changes indicative of myocardial ischemia. Four patients with myocardial infarction and six patients with ischemia had significantly higher than normal inhibition of plasmin activity [100]. The prevalence of hypofibrinolysis was not determined in a control population. In another study, patients < 45 years of age with coronary artery disease (n = 33) had higher concentrations of PAI-1, fibrinogen, von Willebrand factor, and Lp(a) compared to controls (p < 0.05) [101]. Further, plasma fibrin clots in patients were stiffer, contained a greater number of shorter fibrin molecules, and lysed at a slower rate than those found in the control patients. Fibrin stiffness was an independent predictor of premature coronary artery disease and hypofibrinolysis.
The most common coagulopathy is von Willebrand disease (vWD). It results from a quantitative or qualitative defect in von Willebrand factor. The bleeding tendency ranges from mild to severe. Hemophilia A and B are due to deficiencies in coagulation factors VIII and IX, respectively. The tendency for bleeding varies from very mild to severe. The prevalence of atherosclerotic plaques in coagulopathies has been studied by Bilora and colleagues. In a study assessing carotid artery disease in hemophilia and vWD, they found a significantly decreased prevalence of atherosclerotic plaques in coagulopathics compared to controls. By ultrasound, atherosclerotic plaques were present in 13.1% of coagulopathics and 27.2% of controls. 76.3% of all plaques were in controls and 23.6% were in coagulopathics. The degree of stenosis was milder in coagulopathics. Patients with serious coagulopathy had fewer plaques than those with milder disease. 28.9% of coagulopathics were hypertensive, 39.4% were smokers, 36.8% were obese, 17.1% had dyslipidemia, and 6.5% had diabetes mellitus [102].
Subsequently, Bilora and colleagues examined the prevalence of atherosclerotic plaques in the abdominal aorta and lower extremity arteries in 40 patients with hemophilia A and vWD and 40 controls. Plaques were less prevalent in coagulopathics compared to controls. Severe hemophiliacs had fewer plaques than patients with milder disease [103]. In another study examining the carotid, femoral, brachial arteries and abdominal aorta of 50 hemophiliacs and 50 controls, Bilora and colleagues found at least one plaque in six coagulopathics (12.0%) compared to 15 controls (30.0%). 30% of all plaques were in coagulopathics and 70% were in controls [104]. Regarding the coronary circulation in hemophiliacs, Tuinenberg, et al. noted that mortality due to ischemic heart disease was lower in hemophiliacs compared to the general male population. This finding could not be explained by differences in cardiovascular risk factors [105].
The opportunity to study the impact of Factor VIII deficiency on atherogenesis has passed. Prophylactic replacement of factor VIII will ameliorate the bleeding tendency and eliminate any protection from atherothrombosis. This fact was noted in 2009 [105] Results from animal models must be interpreted cautiously. In general, hypercoagulability in mice tends to increase atherosclerosis, whereas hypocoagulability reduces the atherosclerotic burden, depending on the model [106].
Although coagulopathy offers some protection, patients are not completely resistant to atherosclerosis because of the spectrum of disease severity and redundancy in the coagulation cascade and platelet activation, adhesion, and aggregation. For example, factor VII can activate factor X (the "extrinsic" or tissue factor pathway) in the absence of the factor VIIIa-IXa complex. Multiple agonists can activate platelets and cause aggregation, and platelets possess several different receptors which allow them to adhere to multiple substrates.
Myeloproliferative disorders are chronic, clonal proliferation of cells in the bone marrow. Of these, polycythemia vera (PV) and essential polycythemia (ET) are considered to be relatively benign disorders in which arterial and venous thromboses are the main causes of morbidity and mortality [107]. This is reflected in the title of Finazzi, et al.'s 2013 review "Are MPNs [myeloproliferative neoplasia] vascular diseases?" [108]. Blood viscosity is increased in these two disorders, and may become high enough that hyperviscosity syndrome develops. Only older literature on PV is truly revealing about thrombotic complications because of the impact of therapy on reducing red cell mass. Clinical use of radioactive phosphorus began in the late 1930's. Multiple case series and reports from 1926 to 1955 support an increased prevalence of peripheral vascular disease in PV. A 1964 report questioned the association [109]. Regarding ET, Vetrovec, et al. reported that a coronary artery calcification score > 160, indicating high cardiovascular risk, was more common in ET patients than age and sex matched controls with comparable classical risk factors and Framingham risk of coronary heart disease {6/40 vs. 0/42, p < 0.01 [110]. This increased risk was not predicted by the Framingham equation. Keskin, et al. reported the case of a 44-year-old male with ET who suffered acute thrombosis of the celiac artery and had severe atherosclerotic stenoses at the origin of the superior mesenteric artery [111].
Serum viscosity is increased in multiple myeloma. However, the risk of ischemic heart disease is similar to that of age and sex matched controls [112]. This is probably due to the associated anemia of chronic disease, which will tend to normalize blood viscosity. We hypothesize that the anemias of chronic disease and inflammation are compensatory mechanisms which normalize systemic vascular resistance. This response causes the anemias in congestive heart failure [83], monoclonal gammopathies and reactive hypergammaglobulinemias, hemolytic anemias, and others [113].
An effect of statins on thrombosis should not be surprising because pravastatin, atorvastatin, lovastatin, and rosuvastatin therapy decrease blood viscosity [114] and hypercholesterolemia is associated with deep venous thrombosis [53], as noted above. A 2017 meta-analysis showed that statin use reduced the relative risk of deep venous thrombosis by a factor of 0.77 (95% CI: 0.69-0.86, p < 0.0001) [115]. A retrospective study of 152 newly diagnosed systemic lupus erythematosus patients who had no history of previous thrombotic events showed that statin therapy reduced the hazard ratio of thrombosis by a factor of 0.12 (95% CI: 0.01-0.98) [116]. The effect of statin therapy on arterial thrombosis and atherothrombosis are well known. The viscosity lowering effect of statins is not completely explained by their effect on lowering LDL [114].
A recent meta-analysis showed that therapy to lower LDL-cholesterol below 2.59 mmol/L did not provide additional mortality benefit [117]. The impact of LDL on blood viscosity explains the "floor" beyond which further reductions of LDL provide no additional benefits. Figure 6 shows the results of experiments in which the viscosity of erythrocyte suspensions was measured after 24 hours of incubation in varying LDL/HDL ratios [13]. The viscosity of the suspension decreased with decreasing LDL concentration until it reaches a nadir at a ratio of 2.9. Below that ratio, viscosity did not decrease. This suggests that below that ratio, too few receptors are occupied by LDL to foster aggregation. Further reductions of LDL will only decrease plasma viscosity, a weaker determinant of blood viscosity.
 Figure 6: Viscosity of erythrocytes following 24 hours' incubation in serum of various LDL/HDL ratios. Viscosity is expressed as percentage of control, which was erythrocytes suspended in saline, normalized to 100%. The viscosity of the suspension is maximal at an LDL/HDL ratio of 5.7. Viscosity reached a nadir at an LDL/HDL ratio of 2.9. Further reductions of LDL did not decrease the viscosity of the erythrocyte suspension. Modified from ref. [13].
View Figure 6
Figure 6: Viscosity of erythrocytes following 24 hours' incubation in serum of various LDL/HDL ratios. Viscosity is expressed as percentage of control, which was erythrocytes suspended in saline, normalized to 100%. The viscosity of the suspension is maximal at an LDL/HDL ratio of 5.7. Viscosity reached a nadir at an LDL/HDL ratio of 2.9. Further reductions of LDL did not decrease the viscosity of the erythrocyte suspension. Modified from ref. [13].
View Figure 6
Decreased blood viscosity may be one mechanism by which other LDL-lowering therapies such as proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors and ezetimibe reduce the risk of atherothrombosis. It may also be the mechanism by which therapeutic apheresis, blood donation and, in all likelihood, canakinumab, work.
Warfarin inhibits the vitamin K-dependent synthesis of clotting factors II, VII, IX, X, as well as protein C, protein S, and protein Z. The available data suggest that vitamin K antagonists decrease blood viscosity [118,119]. Warfarin prophylaxis prevents both arterial and venous thrombosis. In the Thrombosis Prevention Trial, primary prevention with warfarin to achieve an international normalized ratio (INR) of about 1.5 decreased the incidence of total ischemic heart disease (IHD) events by 21% (95% CI: 4-35, p = 0.02). 1268 males at high risk for IHD aged 45 to 69 years were followed for a total of 18,653 person years [120]. Retrospective review of those data suggested that full compliance with warfarin therapy would have reduced the risk of fatal coronary heart disease by 50% [121]. In the same study, the incidence of stable angina pectoris was reduced by 16%, although this change was not significant (95% CI: -14 to 38, p = 0.26) [122].
Combination oral contraceptives (OC) contain both an estrogen and a progestin, as opposed to oral contraceptives which contain only a progestin. OC are a recognized cause of both venous and arterial thrombosis [123,124]. They are also associated with accelerated atherothrombosis. The risk of thrombotic complications increases synergistically in the presence of other risk factors, particularly smoking, hypertension, and age. The risk decreases with low dose estrogen formulations, which became available in the mid to late 1960's. Conclusions drawn from studies using data collected during the period when high dose estrogen OC were in use can be criticized because of underreporting of adverse reactions, small numbers, incomplete data collection, or, in one study, classifying obese women with those having no predisposing condition to myocardial infarction (MI). In that study, 84 females aged 20 to 44 with no predisposing condition who died of MI in 1966 were analyzed. The incidence of death was higher than expected in OC users and lower than expected in non-users of OCs when compared to the age-matched control population, which was composed of both users and non-users of OC. When the obese were excluded those who died of MI without a predisposing condition, OC users were overrepresented among those who died of MI [125]. The relative risk of thrombotic stroke and myocardial infarction with OC containing 20 μg of ethinyl estradiol changes by a factor of 0.9 to 1.7, and by a factor of 1.3 to 2.3 with OC containing 30 μg to 40 μg with relatively small differences in risk according to progestin type [126].
The Asklepios study showed that OC and hormone replacement therapy (HRT) are associated with accelerated atherogenesis. This study involved a representative sample of the Belgian population age 35-55 without overt cardiovascular disease. Ultrasonography was performed on the carotid and femoral arteries of 1301 women. 27.4% were taking OC and 10% were taking HRT. A history of OC usage for at least one year was present in 81%, with a median usage of 13 years. Multivariate analysis was performed to control for age, smoking, physical activity, blood pressure, lipids, obesity, diabetes, vegetable, fruit, and alcohol consumption, and drug therapy. The odds ratio per 10 years of OC exposure for the presence of atherosclerotic plaque in the carotids was 1.17 (95% CI: 1.00-1.33) and 1.28 (95% CI: 1.10-1.47). The odds ratio for bilateral carotid plaque was 1.42 (95% CI: 1.03-1.84) and 1.34 (95% CI: 1.05-1.63) [127].
Norris and Bonnar reviewed hemostatic changes associated with OC. Both coagulant and fibrinolytic activity are enhanced when taking oral OC. Some subjects also show enhanced platelet activity. Under those conditions, an additional risk factor for thrombosis such as aging or smoking could be sufficient to cause thrombosis [128]. Users of OC have been reported to have shorter thrombin, prothrombin, and partial thromboplastin times, and a hypercoagulable pattern on thromboelastography, consistent with enhanced procoagulant activity.
Most studies have reported high blood viscosity in users of OC because of a higher hematocrit. Lowe and colleagues reported that blood viscosity was higher in users of OC, and that OC usage eliminated the normal gender differences in hematocrit and blood viscosity. Users of OC also had increased fibrinogen levels. They found no difference in blood viscosity or its determinants between female smokers and nonsmokers or between users of OC containing 50 mcg or 30 mcg of estrogen [129]. OC raise triglycerides, which also increases blood viscosity.
The importance of hematocrit in atherothrombosis is shown by the ten year delay in adverse events in females compared to males due in part due to their lower hematocrit as a consequence of menstruation. This difference cannot be explained by plasma lipids or inflammation. Hematocrit is the strongest determinant of blood viscosity and an independent risk factor for coronary artery disease [69]. In the Framingham study, there was an increased risk of cardiovascular morbidity and mortality in subjects with hematocrits in the highest quintile [130]. A 2007 meta-analysis showed that over-treating the anemia of chronic renal disease with erythropoiesis-stimulating agents (ESA) caused increased mortality, arteriovenous shunt thrombosis, and poorly-controlled hypertension [131], all of which are easily attributable to elevated blood viscosity. Because of the increased risk of adverse effects associated with elevated blood viscosity, the U.S. Food and Drug Administration requires that ESA carry a "black box" warning for increasing the risk of myocardial infarction, stroke, venous thromboembolism, and death. ESA should generally be avoided when the hemoglobin concentration exceeds 110 g/L. Determining blood viscosity in patients on ESA should allow a more rational utilization of ESA [114].
Aspirin irreversibly inhibits platelet cyclooxygenase, decreasing production of thromboxane A2 and preventing platelet aggregation by this pathway. It has been shown to be successful for the primary prevention of atherothrombosis. An autopsy study showed a decrease in symptomatic atherosclerosis in arthritis patients with eight or more years of arthritis and aspirin use. In these patients, the prevalence of symptomatic atherosclerosis was inversely related to the duration of arthritis and aspirin therapy [132]. Successful primary prevention in this instance was facilitated by two factors. First, some subjects in this study lived when the prevalence of coronary artery disease in the U.S was higher and died before deaths due to heart disease began to decrease in the late 1960's. Also, most of the patients in this study had rheumatoid arthritis, placing them at increased risk for atherothrombosis. Patients with active rheumatoid arthritis have a greater plasma clot size, faster clot formation, and prolonged fibrinolysis time [133]. In the absence of anemia of chronic inflammation, these patients have elevated blood viscosity due to hypergammaglobulinemia and hyperfibrinogenemia.
In the Physicians' Health Study, aspirin, 325 mg every other day, decreased the risk of myocardial infarction by 44% in 22,071 subjects followed for an average of 60.2 months. Benefit was limited to those aged 50 or older, a group at higher risk for atherothrombosis [134]. Currently, the US Preventive Services Task Force recommends aspirin for the primary prevention of myocardial infarction in men aged 45-79 and the prevention of ischemic stroke in women aged 55-79 when a net benefit is present.
Illicit anabolic steroid use has been associated with cerebral venous sinus thrombosis [135], acute myocardial infarction without underlying coronary atherosclerosis [136], and a greater prevalence of coronary atherosclerotic plaques. Baggish, et al. reported that lifetime anabolic steroid dose was strongly associated with coronary atherosclerotic volume. Total plaque volume increased 0.60 standard deviations units [95% CI: 0.16-1.03 units, p = 0.008] for each additional 10 years of anabolic steroid use [137]. Prothrombotic effects of anabolic steroid abuse include polycythemia [138], dyslipidemia and increased platelet aggregability [139]. Both polycythemia and dyslipidemia increase blood viscosity.
Testing the hypothesis that atherosclerotic plaques develop from the organization of mural thrombi using antithrombotic drugs is complicated because of the risk of bleeding. A better approach would be to study long term intervention with blood donation or therapeutic phlebotomy. These interventions reduce blood viscosity because fresh erythrocytes are more deformable than aged ones. In the Kuopio Ischemic Heart Disease Study, blood donation was associate with a decreased risk of acute myocardial infarction (relative hazard of 0.12, 95% CI 0.02-0.86, p = 0.035) [140]. These results need to be reproduced. Studying blood donation has the advantage of being safe enough to be used as primary prevention in a young population, and could increase the supply of a scarce commodity.
In terms of basic research, much remains to be learned about aortic elasticity. An intervention which slows or prevents progressive aortic stiffening could delay atherothrombosis and increase lifespan. Loss of aortic elasticity is proposed to be one factor which limits lifespan [141].
Disorders and drugs which modulate the risk of thrombosis have a concordant effect on atherogenesis, i.e. the development of an atherosclerotic plaque. This observation supports the hypothesis that atherosclerotic plaques develop from organization of mural thrombi. One process causes both the underlying lesion and its complications, satisfying the Principle of Simplicity (Occam's razor). Many of these entities have no obvious association with lipid abnormalities or inflammation. In fact, absence of the spleen fosters atherothrombosis even though it causes an immunodeficient state. The origin of atherosclerotic plaques from organized mural thrombi is shown by the plaques which develop in synthetic arteriovenous grafts. A common predisposition for mural thrombosis explains why varied risk factors cause the same lesion, the atherosclerotic plaque. The data reviewed here demonstrate the considerable overlap between risk factors for atherothrombosis and deep venous thrombosis.
The authors have no conflicts of interest to disclose.
All authors meet the criteria for authorship as recommended by the International Committee of Medical Journal Editors. They take full responsibility for the scope, direction, content, and editorial decisions relating to the manuscript. All authors contributed equally to the manuscript. All authors reviewed the manuscript.