Cardiovascular Diseases (CVD) are the first cause of death in developed countries, therefore it is of interest to reduce this public health problem. The development of atherosclerosis is the main cause of CVD. This pathology results from the accumulation of lipids in the arterial wall, that leads to a complex inflammatory process. Inflammatory biomarkers are a valuable tool in the detection and monitorization of the evolution of this process, as well as in the choice of therapy to implement. C-Reactive Protein (CRP), determined by high sensitivity methods (hs-CRP), is the most studied biomarker and stands out among the others, being considered an important marker of inflammation. Its importance comes from the fact that its plasma level is not affected by large diurnal or seasonal variations, and for this reason is indicated as an important mediator of the atherosclerotic process.
The goal of this work is to deepen knowledge concerning the importance of C reactive protein as a risk factor for the development of cardiovascular diseases and discuss how it can be used in primary as well as secondary prevention of CVD. The use of C reactive protein as a biomarker allows detection, monitorization and prevention of cardiovascular disease.
Cardiovascular disease, Atherosclerosis, Inflammatory process, Inflammatory biomarkers, High sensitive C reactive protein
Nowadays, Cardiovascular Disorder (CVD) is a health problem with big relevance and concern, being associated with numerous risk factors [1]. For this reason, it is considered one of the most common causes of mortality and morbidity in developed countries. Usually, individuals with CVD only become symptomatic when they reach adult age, however atherosclerosis, the process underlying CVD, frequently starts in childhood with the development of inflammatory processes [2].
Inflammation seems to be crucial in the development and progression of atherosclerosis from the formation of the atheriosclerotic lesion caused by lipid deposition, till rupture of fat plaques [3]. During an inflammatory reaction, there is the development of an interaction between the vascular wall, inflammatory cells and plasma lipoproteins, with the release of several adhesion molecules and cytokines. Together with this, acute phase markers like fibrinogen, C Reactive Protein (CRP), sialic acid, ceruloplasmin, among others are produced [1]. These biomarkers were mentioned in several studies as predictors of cardiovascular disease.
Prevention of cardiovascular disorders must be a priority and all efforts must be done to limit disease progression or avoid the development of a new CVD. In this way, the control of risk factors is a strong weapon in CVD prevention.
Most CVDs result from inadequate lifestyle and hence from controllable risk factors like smoking, sedentarism, excessive weight, bad food diet and stress. However, there are also uncontrollable factors that contribute to the increased risk of CVD like age, family history and gender. Considering gender, last century women were more protected against CVDs than men due to the cardiovascular protection given by estrogens [4]. Nevertheless, nowadays this difference in incidence has been decreasing due to habits acquired by women like the use of oral contraceptives and tobacco.
Other risk factors that might involve genetic and environmental causes are high levels of plasma glucose, since insulin resistance leads to dyslipidemias [5] and increased blood cholesterol levels in the form of LDL (that has higher chance of oxidation and deposition in blood vessels).
Still other CVD risk factors that seem to involve pure genetic causes are high plasma levels of lipoprotein (a) [6], hyperhomocysteinemia [7,8], as well as high levels of C Reactive Protein (CRP) in blood [9].
As mentioned, during the inflammation process there is an increase in acute phase biomarkers like CRP. For this reason, this proteinis of special interest since its plasma concentration is easily quantified, it has the best clinical-epidemiological correlation and, in opposition to other acute phase markers, it shows relatively stable plasma levels, allowing its correct quantification [10]. Besides these findings, this protein shows characteristics that make it very attractive [11]. In the first place, it is an acute phase protein, being an unspecific marker of systemic inflammation [12], whose serum levels increase as a response to several types of lesion, particularly bacterial infections that constitute inflammatory stimuli. Secondly, its hepatic production is mainly stimulated by Interleukin 6 (IL-6) [11]. At the beginning of an inflammatory process, there is an increase in CRP levels in the first eight hours that can reach 300 mg/dL in 48 hours [13]. C reactive protein is an independent risk factor for CVDs [10].
Growing evidences suggest that hs-CRP constitutes an important cardiovascular risk marker and is physio-pathologically associated with the atherosclerotic process, having value in primary and secondary prevention [11]. However, its serum concentrations can be influenced by other factors like pharmaceutical products, hormones and tobacco [12].
Atherosclerosis starts with systemic mediators that have a key role in its progression, as well as in its stability and plaque rupture. The association between inflammatory biomarkers, the atherosclerotic phase and the dynamic of cardiovascular events related to atherosclerosis seems to be fundamental for CVD development [14]. Chronic inflammation of blood vessels seems to be the main cause of atherosclerotic plaque formation and its rupture [15]. Plaque formation results from accumulation of lipids, inflammatory cells and fibrotic elements, that deposit in the wall of arteries and blood vessels, leading to their obstruction [16].
The inflammatory process initiates with the increase of endothelium permeability to inflammatory cells, giving rise to oxidized LDL deposition [17]. After endothelium dysfunction, mononuclear cells like monocytes and T lymphocytes adhere firmly to the endothelium and migrate to involving tissues (sub-endothelial space) by a process called diapedesis. The attachment of leucocytes to endothelium is activated by adhesion molecules like Selectins (selectin-E, selectin-P), cellular adhesion molecules (Inter-Cellular Adhesion Molecule-1 (ICAM-1), Vascular Adhesion Molecule-1 (VCAM-1)) and integrins. Chemotaxis and monocytes entrance to the sub-endothelial space is promoted by a Protein (protein-1) that potentiates attraction of monocytes, Interleukin-8 (IL-8), and one chemokine, called fractalkine. After that, monocytes and macrophages start to differentiate. Macrophages receive oxidised LDL via scavenger receptor (CD36, scavenger receptor-A), becoming sponge cells, lesion indicators [1]. Later, smooth muscle cells migrate into the intima, proliferate and forma fibrous matrix, that keeps on growing, narrowing the artery.
It is believed that, during the processes of necrosis and apoptosis, lipid filled macrophages release matrix metalloproteinases, that damage the endothelium. Since LDL filled macrophages are enriched in tissue factor, this one is released from the macrophage and contacts circulating platelets, resulting in the formation of thrombi and consequent acute coronary syndromes [1].
CRP was first described by Tillet and Francis, more than 70 years ago, during a study in patients having infection with Streptococcus pneumoniae. Serum obtained from these patients, during the acute phase of symptoms, included a substance capable of precipitating the C polysaccharide of the cell wall of pneumococcus. This substance was called C reactive. Several studies showed that this protein is undetectable in healthy patients, however reaches very high levels in patients having infections. After patient's recovery, levels of this protein become undetectable again [18].
CRP is a member of the family of proteins called pentraxins and has a molecular weight of 115135 Da. This protein is formed by five identical non-glycosylated polypeptide chains (23027 Da) of 206 aminoacids each. The semonomers are linked in a non-covalent fashion, organized in a very stable discoid structure with resistance to proteolysis [19]. Each monomer has two distinct faces, the recognition/binding face and the effector face. The binding face, to which two calcium ions are associated, recognises the phosphocholine residues of the C polysaccharide of Streptococcus pneumoniae [20,21], and the effector face has affinity for the complement factor 1q and FcγRs [20].
CRP is an acute phase protein produced in liver due to stimulation by several pro-inflammatory cytokines derived from monocytes/macrophages or adipose tissue [11,22]. Its liver production is mainly stimulated by IL-6, being this synthesis synergically increased by IL-1 [18,23]. It is consensus that pro-inflammatory risk factors like oxidized LDL and infectious agents such as Chlamydia pneumoniae trigger a pro-inflammatory response [24]. This response leads to the increase in IL-1β and TNF-α secretion, that consequently provokes release of cytokine IL-6. More over, IL-6, after binding to its receptor in liver, leads to secretion and release of CRP and amyloid A serum protein [22].
Besides liver production, data suggests that CRP is also produced in the atherosclerotic lesion (specially in smooth muscle and macrophages), in kidneys, neurons and alveolar macrophages [25,26]. Moreover, there are evidences of stimulation of CRP production by lipid peroxidation and infection by cytomegalovirus that triggers a cascade of pro-inflammatory cytokines [3].
Work of Chang, et al. [27] showed that CRP does not bind to native LDL. Instead, the increase in reactive oxygen production leads to oxidation of LDL that triggers entrance of LDL in macrophages mediated by CRP. CRP binds to oxidized phosphatidylcholine present at the surface of LDL [27].
Healthy individuals have low plasma levels of CRP (< 0.5 mg/dL). However, during inflammatory processes, serum concentration of this protein can rise one hundred to one thousand times. Its synthesis starts 4-6 hours after stimulation, duplicates every 8 hours and reaches a maximum after 50 hours (around 2 days). Its levels can reach 300 mg/dL in 48 hours. These characteristics make CRP a relevant clinical marker due to its stability, high sensitivity, good reproductivity and precision. This protein has a plasma half-life of 19 hours even after stimulus, trauma or surgery. It can take several days until it reaches basal levels [28,29]. In the absence of chronic stimulus, CRP levels normalize in 3-4 days. On the other hand, in chronic inflammatory states, CRP concentrations can remain high forever [28].
It has also been observed a correlation between CRP genetic variants and its plasma levels. Michopoulos, et al. [30] saw increased CRP plasma levels associated with the presence of the rs1130864 CRP polymorphism. Moreover, Markt, et al. [31] detected mean CRP levels significantly elevated in men with one copy of the variant alleles rs3093075 and rs1417938 compared to those with no copies. On the other hand, the same study observed that men with one copy of the variant allele rs1800947 had significantly lower mean CRP levels. The rs2808630 polymorphism was not associated with circulating CRP [31].
CRP plasma concentration differs between individuals. Each person has its own basal level that remains stable over the years without big changes during the day or season. Diet does not interfere with CRP levels as well. However, these values tend to increase slightly with age in healthy individuals, probably due to the development of diseases still in the sub-clinical stages [29].
Between all existing markers, CRP shows up due to its high sensitivity in evaluating inflammatory states, showing a strong increase as a response to several inflammatory and infectious stimuli. Measurement of CRP plasma levels helps the clinical evaluation of the presence, extension and activity of the inflammatory process and the follow-up of the evolution and therapeutic response [28].
In the beginning, CRP plasma levels were determined by serum-agglutination performed in latex particles in a slide. This assay allowed a semi-quantitative determination, with a subjective interpretation. Later, quantitative methods for CRP quantification were developed through immunoturbidimetry and nephelometry [32].
Due to the low sensitivity of these methods on detection of low levels of inflammation, in the 90s a high sensitivity nephelometry method for detection of very low levels of serum CRP was developed, called High Sensitivity CRP (hs-CRP). This is the ideal laboratory method in use to determine cardiovascular risk linked with chronic systemic vascular inflammation of atherosclerosis, having a detection limit of 0.3 mg/L [11].
Table 1 describes the relationship between hs-CRP plasma concentrations and the risk factor, or capacity of cardiovascular events prediction.
Table 1: Risk degree in the development of cardiovascular problems related to serum levels of hs-CRP [1]. View Table 1
Several studies confirm that hs-CRP predicts cardiovascular events in healthy women [33] and men [34], in individuals with traditional risk factors [35] or with CVD [23]. The capacity of hs-CRP to predict CVD risk is independent from traditional risk factors [34,36]. hs-CRP is an additional risk factor for coronary artery disease when associated with high plasma levels of cholesterol [22,37].
For many years, CRP was used as a complementary method for diagnosis of inflammatory processes of any nature. Meanwhile, with the discovery of inflammatory components involved in cardiovascular events, mainly atherosclerosis, CRP was assigned as a risk indicator for coronary disease and cerebral vascular accidents [28], being an inflammatory marker considered strong predictor, that is independent of the risk for cardiovascular event or death.
Several prospective clinical studies have shown that CRP is associated with mortality risk in short or long term both in patients having acute or chronic ischemic cardiac disease, or in individuals with atherosclerosis risk [38]. In 2003, Ishikawa, et al. [39] concluded that CRP is located inside the atherosclerotic plaque, having an important role in plaque vulnerability. In the same way Inoue, et al. [24] in 2005, showed that CRP is produced in the atherosclerotic plaque responsible for acute coronary syndrome, demonstrating the existence of a CRP gradient in coronary arterial circulation close and far away from the plaque.
Evidences suggest that CRP is not only an inflammatory marker, but also participates actively in the atherosclerosis process [40]. Its levels correlate directly with several cardiovascular risk factors, like body mass index, smoking, systolic arterial pressure, serum levels of triglycerides and total cholesterol, cardiac frequency, fasting glycaemia and cardiovascular disease or stroke history and in an inverse way with HDL levels, both in children and in adults [41,42].
By its interaction with classic risk factors, CRP can lead to changes in the pro-atherosclerotic profile of patients [43]. This protein has direct influence in atherosclerotic vessels through the activation of the complement system, closely related with the initial stages of atherosclerotic plaque formation, and with the stimulus of tissue factor synthesis by monocytes (procoagulant effect), and consequently, in promotion of inflammation and thrombosis [10].
CRP favours the development of a pro-inflammatory state, through reduction of endothelial Nitric Oxide Synthase Activity (eNOS) [29], as well as reduction of transcription of eNOS coding genein endothelial cells [29].
Nitric oxide produced by endothelial cells has an essential role in blood vessels relaxation, being strategically linked to the endothelial cell membrane. Moreover, NO contradicts vascular smooth muscle contraction and inhibits platelet activation. It also acts on integrins, by modifying leucocyte adhesion and neutrophil diapedeses [44]. NO is continuously released from vascular endothelium, being responsible for the maintenance of tissues blood flow and for tissue extravasation control [45].
Therefore, all described processes are limited when CRP plasma levels are high [29]. By inhibiting NO production, CRP contributes to apoptosis of endothelial cells and consequently to pro-atherogenic and pro-inflammatory events (Figure 1) [46].
 Figure 1: Cascade of changes induced by CRP that lead to cardiovascular events [46]. View Figure 1
Figure 1: Cascade of changes induced by CRP that lead to cardiovascular events [46]. View Figure 1
Additionally, CRP not only controls the expression of adhesion molecules, like ICAM-1 and VCAM-1, through chemokine MCP-1, but also facilitates entrance of LDL, by opsonization, in macrophages [29,47]. Moreover, it has the power to induce oxidation of that cholesterol, contributing for the development of atherosclerosis. CRP is also involved in destabilization of atheroma fibrous layer, by stimulating Matrix Methaloproteinase-1 (MMP-1), released with collagen and proteins degradation. This destabilization occurs equally by the decrease in concentration of Plasminogen Activator (tPA), responsible for clot lysis in vascular wall, and for the increase in the level of Plasminogen Activator Inhibitor (PAI-1), that inhibits fibrinolysis. This process facilitates thrombi formation in endothelial wall, increasing the risk of cardiovascular events [48,49] (Figure 2).
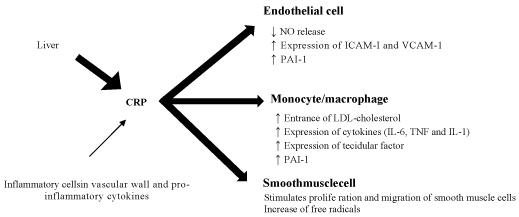 Figure 2: CRP role in stimulation of endothelial cells, mononuclear cells (monocytes and macrophages) and smooth muscle cells leading to the production of inflammatory mediators [49]. View Figure 2
Figure 2: CRP role in stimulation of endothelial cells, mononuclear cells (monocytes and macrophages) and smooth muscle cells leading to the production of inflammatory mediators [49]. View Figure 2
Therefore, CRP is not only an inflammatory marker of atherosclerosis and coronary events, but also a CVD mediator due to its contribution for the lesion process, for plaque break and incoronary thrombosis mechanisms [29,46]. However, more research is needed to be able to conclude about the effects of CRP as a risk factor of CVD and its clinical relevance [47].
Due to the relationship between high CRP plasma levels and cardiovascular mortality and morbidity risk [11], it is important to establish a primary care line to decrease CVDs. For this, it is essential the evaluation of cardiovascular risk factors to stop their progression.
Several prospective studies having CRP as central target, have shown the benefits of primary prevention. In 1999, the MONICA-Augsburg study [50] performed in a sample of 936 asymptomatic men, concluded that the increase of hs-CRP leads to a 19% increased risk of fatal and non-fatal coronary events.
In the same way, the PREVEND study in 8139 asymptomatic men and women observed a relationship between hs-CRP and angiographic characteristics and consequently clinical instability of the atherosclerotic plaque [51]. The use of lovastatin in the treatment of 5742 individuals (AFCAPS/TexCAPS study) reduced the occurrence of the first coronary event both in individuals with high cholesterol levels, and in the ones having high hs-CRP levels, even with low lipid profile [52].
The PRINCE study [53] performed with 1702 asymptomatic men and women showed that pravastatin reduces hs-CRP levels in individuals without previous history of CVD, independently of cholesterol levels.
Rosuvastatin was tested in 1802 asymptomatic men and women (having cholesterol LDL < 130 mg/dL and hs-CRP > 2 mg/L) in the JUPITER study [54]. It was observed a reduction of 50% in LDL levels and 37% in hs-CRP levels, consequently reducing cardiovascular events in these apparently healthy individuals.
Described data show that the use of statins in primary prevention of CVD leads to a decrease in CVD mortality. It was also seen the decrease of coronary events in groups with different characteristics of the population under study [54].
According with guidelines concerning "Emerging biomarkers for primary prevention of cardiovascular disease" from the National Academy of Clinical Biochemistry Laboratory [55], hs-CRP cannot be determined in general population to evaluate cardiovascular risk. However, it can be used to stratify cardiovascular risk in adults with intermediate risk of coronary disease, and can help the decision about statins use in primary prevention [11].
The objectives of secondary prevention are focused in avoiding a new cardiovascular event and identify the progression of myocardium dysfunction, leading to a reduction of mortality rate due to these diseases. To achieve these goals, secondary prevention includes changes in lifestyle and continuous use of medicines [56].
For hypertensive patients it is essential smoking cessation, weight loss, decrease of cholesterol serum levels and practice of physical exercise. Moreover, the continuous use of aspirin, β-blockers, inhibitors of the angiotensin I conversion enzyme, and in women in menopause, hormone replacement are also essential [56].
Several studies have shown the value of CRP in secondary prevention of CVDs. The FRISC study [57] performed in 917 patients with acute coronary syndrome showed that hs-CRP has independent capacity of cardiovascular mortality prediction. Another randomized prospective study in 391 patients having non-fatal/fatal acute myocardial infarction (CARE study [36]), proved that the use of pravastatin leads to risk reduction of coronary events, mainly in individuals with inflammation evidence determined by hs-CRP, not related with lipid levels.
Intensive therapy with a statin applied to 3745 patients with acute coronary syndrome (PROVE-IT study [58]) could reduce hs-CRP levels below 2 mg/L, consequently decreasing the risk of acute myocardial infarction or fatal coronary events, independently on the reduction of LDL levels.
The PEACE study [59] (with 3771 individuals havingstable coronary disease) allowed to associate plasma hs-CRP levels above 1 mg/L, to higher mortality risk due to cardiovascular disease, acute myocardial infarction or stroke.
Since the inflammatory process is an integral part of atherosclerosis evolution, the use of a biomarker like CRP, becomes quite useful in combination with the control of classic risk factors like lipid levels, change of eating habits, weight loss associated with regular physical activity, control of glycaemic levels and smoking cessation. The interconnection of these factors are strategies for reduction of cardiovascular events in primary and secondary prevention [29,47].
Knowing that CRP is involved in CVD pathophysiology, it is expected that reduction of CRP levels prevents the development of the disorder and its complications. Data suggests that certain drugs can reduce CRP levels, however, the clinical relevance of these interventions is not known [60].
Prevention of cardiovascular events in individuals with high CRP levels can be achieved using CRP lowering agents. Table 2 summarizes the main studied agents/strategies that contribute to reduction of plasma CRP.
Table 2: CRP lowering agents. View Table 2
Aspirin has the capacity of reducing the risk offuture cardiovascular events due to its antiplatelet and anti-inflammatory properties [47,60]. In 1997, Ridker and collaborators [34] published the first study that demonstrated the effect on reduction of cardiovascular events, because of therapy with acetylsalicylic acid, together with serum CRP levels reduction [34]. This study showed that a dose of aspirin per day contributes to a 44% decrease in myocardial infarction risk. Patients having higher CRP levels showed higher risk decrease [34]. In the same way, a 75 mg per day dosage during a month led to a meaningful reduction in plasma CRP [61]. On the other hand, other studies demonstrated the ineffectiveness of aspirin relative to CRP levels [62]. Cyclooxygenase-2 Inhibitors (COX-2) (rofecoxib, celecoxib) were also tested for their ability to reduce CRP levels. Some studies observed a decrease on serum CRP levels, when used alone or in combination with statins or aspirin [63,64].
In the same way, Lincoff, et al. [65] tested the effect of clopidogrel, an antiplatelet agent,on CRP levels and observed a 32% reduction [65].
Besides the anti-inflammatory effects on atherosclerotic tissues, statins also lead to reduction in CRP levels [53]. Riesen, et al. [66] observed beneficial effects on plasma CRP reduction. In this study, it was observed that therapy with high dosage of statins or its combination with COX-2 inhibitors, given before an invasive coronary procedure in patients with unstable angina, rapidly decreased CRP serum levels [66].
There are, in fact, numerous studies showing the power of statins in reducing hs-CRP levels, that observed a reduction of 13 to 50% in its plasma levels [37-70]. This was observed in patients with obesity [68], diabetes [70], dyslipidemias [67,68] and coronary artery disease [69]. This statin associated with hs-CRP levels reduction results in a bigger clinical benefit in the treatment of patients having atrial fibrillation [71].
Another lipid lowering agent, ezetimibe, an inhibitor of cholesterol absorption, was tested in combination with statins [67]. This study observed a 41% reduction in CRP plasma levels when using the combined treatment [67]. Fenofibrate, usually used in the treatment of hypertriglyceridemia also led to CRP reduction, being observed a decrease between 74-84% [72,73]. In the same way, treatment of dyslipidemias with a combination of niacin/statin, allowed a 24% reduction of CRP [74,75]. The effect of niacin alone was not tested in these studies.
Some antidiabetic agents can also have beneficial effects on CRP levels. A 40% reduction of CRP levels, after treatment with thiazolidinedione derivatives, was observed in type 2 diabetes mellitus patients [76] and in nondiabetic hypertensive patients [77]. Moreover, β-adrenoreceptor antagonists, normally use in the treatment of hypertension and coronary artery disease, led to reduction of CRP levels ranging between 36-73%, depending on the type of antagonist used [78-81].
The use of antioxidants has also been shown to contribute to CRP reduction. Several studies observed decreased plasma CRP levels when using vitamin E, ranging from 49 to 81% [82-85], but no effect was seen by vitamin C [86].
Another group of drugs known to have effect on CRP plasma levels are the inhibitors of Renin-Angiotensin system. Angiotensin converting enzyme inhibitors and angiotensin II receptor blockers were tested and both led to decrease in CRP levels [87,88].
Some calcium channel antagonists can also lead to plasma CRP reduction. This was observed in the study of Hung, et al. [89] where these agents were used in the treatment of patients with vasospastic coronary artery disease.
The use of herbal extracts like curcuminoids, multifunctional natural products with promising cardioprotective and anti-inflammatory properties, has also been tested. Sahebkar [90] observed a significant decrease of CRP plasma levels in a group of 172 individuals.
Moreover, obesity, tobacco, and lack of physical activity are factors associated with high levels of CRP, and therefore changes in lifestyle also contribute to the intended reduction [10,47]. One example is the use of lipid lowering diets tested by Jenkins, et al. [91], who observed a 10-28% decrease in serum levels of that protein, depending on the type of diet used.
Lack of physical activity during childhood and adolescence is a risk factor with big significance for CVD [92]. Regular physical exercise is associated with potential benefits for health, being considered essential for proper growth and development [93]. Data proved that regular physical activity is inversely associated with high levels of different inflammatory markers [94-96]. It has also been observed that the type, duration and intensity of physical activity are crucial factors for the profile of cytokine response after exercise [97]. A study of Donges, et al. [98] evaluated changes of IL-6 and CRP after 10 weeks of resistance and aerobic physical exercise in sedentary adults. This studydid not show significative reductions of CRP and IL-6 after aerobic training, however, reductions of CRP plasma levels were observed in response to resistance training [98]. Smith and co-workers [99] have shown that long term physical exercise can strongly reduce plasma levels of inflammatory cytokines and CRP. This anti-inflammatory effect demonstrates the benefits of physical activity in cardiovascular disease prevention [99].
As mentioned previously, CRP has a long half-life (around 19 hours), reaching its basal levels after several days. Indeed, the prognostic value and the evolution of the treatment in an initial phase might not be the most correct, making it a limitation on its use. Moreover, CRP takes 4-6 hours to be produced, after stimulus. So, its detection might not be fast enough to detect a cardiovascular event [28].
CRP is an unspecific marker of acute phase response and increases of its plasma levels are expected not only in inflammation, but also in other clinical situations like infection and necrosis [100,101]. In this way, it is important to say that CRP like any other marker, cannot be used alone in diagnosis or monitorization of therapeutic response.
CVDs are the main cause of incapacity and premature death in the whole world. In these circulatory diseases, atherosclerosis is the basis for their progression. However, CVD development does not depend on one factor alone, but on the association of several risk factors, resulting in a synergetic and multiplicative effect. With the high prevalence of risk factors for CVD in the general population, it becomes essential to give special attention to their prevention, detection and correction.
CVD prevention should start during childhood and adolescence with the adoption of healthy habits, since these will be used in adult life. Risk of CVD development can be detected using biomarkers that have been relevant in diagnosis, prognosis and as a therapeutic guide for these disorders. Biomarkers constitute a biological-qualitative and quantitative parameter of physiological changes or pathological processes.
Among biomarkers, CRP, an inflammatory marker, is considered to have bigger clinical relevanceand higher additional prognostic information, being independent of traditional risk factors. Moreover, CRP is an inflammatory mediator in cardiovascular events.
CRP production, together with the increase of other pro-inflammatory cytokines starts during the first stage of the inflammatory process. This shows its role as a risk marker in this process. Moreover, CRP leads to instability of vascular endothelium by reducing NO production, induces LDL oxidation by facilitating LDL entrance in the macrophage, and is involved in destabilization of the fibrous layer of the atheroma. This destabilization occurs through the stimulus of matrix methaloproteinase-1, released with collagen and other proteins degradation, inducing thrombus formation in the endothelial wall and consequently increasing the risk of cardiovascular events.
So, CRP measured by high sensitivity methods in individuals having apparent cardiovascular risk, should be seen not only as an inflammation marker but also as a possible risk factor for CVDs.
High CRP levels can be reduced by the use of several strategies that go from changes in daily habits to the use of several drugs that directly interfere with CRP serum concentration.
In conclusion, the prevalence of high risk individuals for the progression of CVD, points out for the need of strategies with coordinate intervention of several health experts. These strategies should involve prevention, detection and effective treatment of cardiovascular risk factors, contributing in this way to the risk reduction in the population.
It is important the adoption of measures in the population, that potentiate the reduction of the risk of developing this type of disorders, the application of a fast and adequate treatment and the use of secondary prevention measures that will reduce their return.
The Authors declare that there is no conflict of interest.
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.