The main aim of this case report was to present the method of diagnosis, management, and prognosis of a young adolescent diagnosed with a desmoplastic small round cell tumor (DSRCT).
We report a new case of a 16-year-old female who proceeded complaining about non-specific abdominal pain. Physical examination revealed a palpable mass in the lower abdomen and a diffuse abdominal pain. Abdominal ultrasonography showed an irregular pelvic mass, with heterogeneous echo structure making 12 cm × 8 cm. She underwent an exploratory laparotomy and the excision of the mass revealed desmoplastic small round cell tumor. She died 6 months after diagnosis despite chemotherapy.
Although a DSRCT can develop at various sites, the intra-abdominal site is the most common location. Due to our limited knowledge of the pathologic and clinical nature of this disease, there is no clear consensus regarding the optimal therapeutic procedures for treating this neoplasm and a multimodal treatment with surgery, chemotherapy and radiotherapy is used for these rare cases. A high degree of care and improvements in diagnostic capabilities are required in order to identify this entity and avoid misdiagnosis.
Desmoplastic small round cell tumor, Peritoneal carcinomatosis, Multimodal therapy, Chemotherapy, Radiation therapy
Desmoplastic small round cell tumor (DSRCT) is a rare and highly aggressive mesenchymal tumor; fewer than 200 cases have been reported in the literature [1]. DSRCT mainly develops in adolescent and young adults with a strong male predominance; the male to female ratio is 4:1 [2]. Among female, it appears more precociously in the form of a pelvic mass, an abdominal distension or a high rate of Ca125. Its diagnosis is anatomopathological. The tumor typically develops in the abdominal cavity, invading the omentum with multiple peritoneal implants involving the diaphragm, splenic hilum, mesentery of small and large bowel, and the pelvic peritoneum. This tumor has very bad prognosis which can evolve on the metastatic mode [3]. As for of others intraabdominal tumors, patients may be asymptomatic for long periods of time and diagnosis is made when tumor burden is large. Clinically, patients present symptoms of abdominal sarcomatosis such as ascites, abdominal pain and/or distension, constipation or bowel obstruction, vomiting, and weight loss [1].
We report a case of desmoplastic small round cell tumor occurring in a 16-year-old young girl miming an ovarian tumor.
Miss M.S., aged 16, with no history pathological individuals, was admitted in August 2011 for exploration of an abdominal mass. The onset of her illness was in one month by installing abdominal pain type of gravity and a gradual increase in the volume of the abdomen. She had no gynaecological, urinary or other gastrointestinal symptoms. The clinical examination revealed a pelvic mass arriving to the umbilicus, mobile, well-circumscribed, painless making about 12 cm. The abdominopelvic ultrasound found an irregular pelvic mass, with heterogeneous echo structure making 12 cm × 8 cm. The tumor is highly vascularized in the color Doppler (Figure 1). The patient underwent a laparotomy revealed a tumor developed depends on the greater omentum, very vascularised. The exploration of the abdominal cavity does not find any abnormality. An ommentectomy carrying a tumor was performed (Figure 2). The result of the pathological study was in favor of a desmoplastic round cell tumor (Figure 3). Immunohistochemical study showed an expression by tumor cells of the EMA, desmin, vimentin, WT1 and S100 protein. The study by RT-PCR did not detected the specific transcript of the translocation t(11,22) (p13, q12) [EWS/WT1]. The staging showed no secondary location.
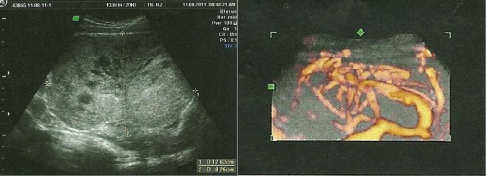 Figure 1: Ultrasound images of the tumor highlighting the heterogeneity and hypervascularisation at color Doppler.
View Figure 1
Figure 1: Ultrasound images of the tumor highlighting the heterogeneity and hypervascularisation at color Doppler.
View Figure 1
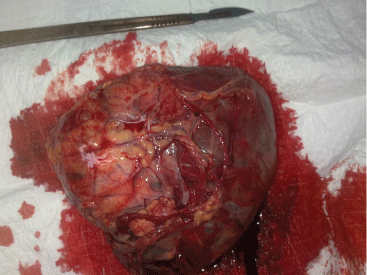 Figure 2: Macroscopic aspect: Tumor depends on the peritoneum having a consistency richly vascularized.
View Figure 2
Figure 2: Macroscopic aspect: Tumor depends on the peritoneum having a consistency richly vascularized.
View Figure 2
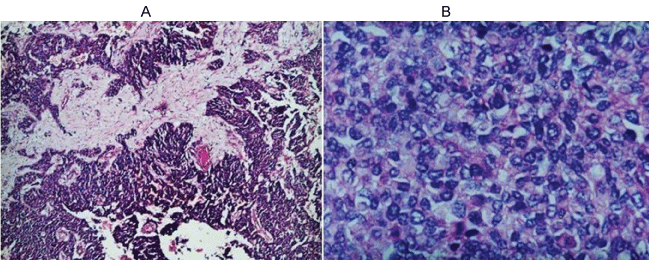 Figure 3: Morphological appearance A) Small round blue cells arranged in columns, and span and pseudo-rosettes in desmoplastic stroma; B) Tumor cells are monomorphic to anisocaryotiques round nuclei showing mitotic figures.
View Figure 3
Figure 3: Morphological appearance A) Small round blue cells arranged in columns, and span and pseudo-rosettes in desmoplastic stroma; B) Tumor cells are monomorphic to anisocaryotiques round nuclei showing mitotic figures.
View Figure 3
Additional treatment with three cycles of chemotherapy was started involving doxorubicin and ifosfamide associated with granocyte® (G CSF: Granulocyte-colony-stimulating factor) which reduces the toxicity of chemotherapy. The evolution is characterized by pelvic recurrence after two cycles of chemotherapy confirmed by the scanner as a left adnexal pelvic mass by 5 cm associated with iliac lymph nodes (Figure 4A).
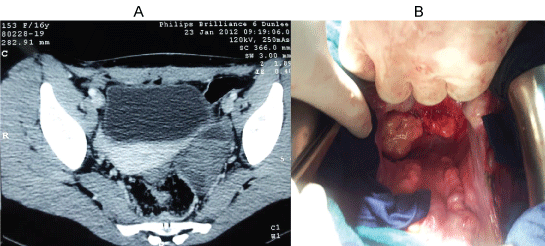 Figure 4: A) Abdominal pelvic CT showing a left latero-uterine mass making 53 × 33 mm with bilateral iliac lymph nodes; B) Intraoperative appearance of tumor masses at the Douglas.
View Figure 4
Figure 4: A) Abdominal pelvic CT showing a left latero-uterine mass making 53 × 33 mm with bilateral iliac lymph nodes; B) Intraoperative appearance of tumor masses at the Douglas.
View Figure 4
Reoperation finds two tumor masses at the Douglas (Figure 4B); the most voluminous is in intimate contact with the front of the rectum, with nodules in the appendix, peritoneal nodules, and bilateral iliac lymph nodes. The other organs were not affected, including the liver, the uterus and its annexes. We conducted a gesture with palliative resection of tumor masses, a bilateral ilio-obturator lymphadenectomy and appendectomy. Histological examination confirmed the recurrence of the same tumor, in addition to the detection of specific transcript t (11; 22) (p13; q12) in the cytogenetic study.
The patient received two cycles of chemotherapy involving docetaxel, gemcitabine and cyclophosphamide but she unfortunately experienced progressive disease and started a palliative treatment by the tyrosine kinase inhibitor imatinib (Gleevec®). She expired 6 months after her initial diagnosis following the deterioration of its general condition.
Desmoplastic Small Round Cell Tumour (DSRCT) was first described in 1989 [4]. DSRCT is a rare and highly aggressive tumour that usually occurs in males during adolescence and early adulthood.
The clinical pattern of disease is typically one of multiple intra-abdominal tumour deposits on serosal surfaces.
Primary DSRCTs have been described originating outside the abdominal cavity arising from the other mesothelial surfaces such as lung pleura and tunica vaginalis [5]. DSRCT can also arise from solid organs such as the ovaries, liver, kidneys, pancreas, bone and even the posterior cranial fossa [6].
Patients typically present with pain, palpable mass, abdominal distension, ascite or symptoms related to obstruction of viscus [2,7].
The most frequent sites of metastases are liver, lymph nodes, lung, and bone marrow [8]. However, the invasion of adjacent organs is rare [9].
Radiologically, DSRCT is similar to the other intraabdominal primary tumours. Abdominal imaging by ultrasound, computed tomography scan or magnetic resonance imaging reveal unique or multiple peritoneal masses.
In our observation, abdominopelvic ultrasound was sufficient, but a complement CT was performed postoperatively after histological result.
The diagnosis is made after histological and immunohistochemical study of the resected tumor or biopsy during laparoscopy, however cytogenetic study requires samples of good quality [10].
Macroscopically, the tumor comes in the form of single or multiple nodules of variable size, developed at surface of the peritoneum. Sometimes, tumoral element is dominant surrounded satellite nodules on peritoneum. The tumor has a firm consistency, smooth and bumpy surface. The cut surface is grayish white with foci of necrosis sometimes associated with myxoid areas.
Histologically, DSRCT have large histological polymorphism, the most usual appearance is that of a small round cell proliferation, undifferentiated, monomorphic, dispersed in a generally abundant desmoplastic stroma [11]. Other morphological variants are described, involving rhabdoid, clear, pleomorphic, basaloid and glandular areas [12]. These atypical forms make the diagnosis particularly difficult, especially as desmoplasia may be focal or absent [12].
The immunohistochemical profile is characterized by the co-expression of epithelial (cytokeratin, EMA), mesenchymal (vimentin, desmin) and neural markers (NSE). The positivity of desmin reached 95% of cases, the epithelial markers and vimentin concerns 90% of cases and NSE approximately 75% of cases [13]. Immunohistochemical phenotype may be incomplete making the diagnosis particularly difficult.
The EWS-WT1 gene fusion has more recently been discovered to be disease-specific to DSRCT. This characteristic reciprocal translocation is demonstrated by fluorescence in situ hybridization (FISH) or reverse transcriptase-polymerase chain reaction technique and is specific to DSRCT at any primary location. It is the definitive diagnostic marker [4].
Due to the limited knowledge on the pathologic and clinical nature of this disease, there is no clear consensus regarding the optimal therapeutic procedures for treating this neoplasm. However, a multidisciplinary approach is required. It combines:
- Total resection of the mass and postoperative multiagent chemotherapy and radiation therapy as the best therapeutic options. Total resection is not often feasible because of the numerous peritoneal implants [7].
- Preoperative chemotherapy, with the P6 protocol including cyclophosphamide, doxorubicin, vincristine, ifosfamide and etoposide, as a way of shrinking the widespread mass and reducing its vascularity [7].
- Adjuvant radiotherapy to the whole abdominopelvic area was also delivered.
The prognosis of DSRCT is very reserved, despite aggressive treatment, with an average survival of less than 30 months and only 44% and 15% of patients alive at 3 and 5 years respectively after diagnosis [8]. Patients with liver metastases have a poor prognosis with an average survival of 18 months [8].
Other therapeutic methods, citing immunotherapy, inhibitor of protein tyrosine kinase or Imatinib (Gleevec ®), don't give positive answers but are still being tested [8]. Intraperitoneal hyperthermic chemoperfusion has also been administered as an effort to reduce the bulk of the tumor [3].
Despite its rare incidence in women, the diagnosis of DSRCT must be considered in a young woman presenting abdominopelvic symptoms and one or more peritoneal masses.
The DSRCT are characterized by a large histological polymorphism, and immunohistochemical studies and cytogenetic or molecular biology are necessary for diagnosis. The prognosis remains bleak and evolution is usually done to death, despite a heavy therapeutic care including surgery, high-dose chemotherapy and sometimes radiotherapy.
The authors have no conflicts of interest relevant to this article.