Over forty years of extensive research has not yet solved the complexity of Ebola pathogenesis leading to a continued need for a successful cure. This virus has evolved different strategies to counteract immune responses as they are unreceptive to a large portion of the known antiviral drugs and there is no valid treatment as to date for disease created by this pathogen. A plethora of evidences have revealed that monocytes/macrophage contribute significantly in viral pathogenesis.
To interpret how macrophages and effector molecules respond against Zaire Ebola virus infections we evaluated the expression of genes that typically distinguish M0 and M1/M2 subsets by microarray dataset analysis.
We identified differentially expressed genes including in peripheral blood mononuclear cells (PBMCs) of Ebola infected Cynemologus macaques employing the reference gene expression dataset GSE5099 obtained from the NCBI Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) database.
Microarray analysis showed that the 3th and 4th days post infection was characterized by a massive presence of M1/MΦs as indicated by the expression of pro-inflammatory mediators. Nevertheless, a significant presence of M2 was observed until the 5th day post infection. Moreover, we observed a gradual evolution in the gene expression patterns of markers such as TLRs, OASs, HLA and Chitinase-like-lectins over time from day 0 to day 6, usually with a more significant change at 4th day, resulting in the separation of the early acute (day 0 to day 3) and late acute phases (day 3 to day 6).
Collectively, this revised analysis provided new avenues of research on Ebola pathogenesis and persistence contributing for the future development of more effective anti-Ebola diagnostic and therapeutic interventions that may pave the way to viral eradication.
Macrophages, ZEBOV, OASs, TLRs, HLA, Chitinases
ZEBOV: Zaire Ebola Virus; MΦ: Macrophages; OASs: 2',5'-Oligoadenylate Synthase; TLRs: Tool Like Receptor; HLA: Human Leukocyte Antigen; CHI3L1: Chitinase-Like 1 Proteins; CHI3L2: Chitinase-Like 2 Proteins; CHID1 or SI-CLP: Stabilin-Interacting Chitinase-Like Proteins
Ebola virus is the etiologic agent of epidemic severe haemorrhagic fever in humans with a high mortality rate and a huge economic impact on resource-poor settings [1]. The Zaire Ebola virus (ZEBOV), along with four other Ebola species (Sudan, Ivory Coast, Bundibugyo and Reston) belong to the Filoviridae family. The species ZEBOV shows the highest mortality in humans (~90%) and non-human primates [2]. Filoviruses may enter through minor lesions on the skin and subsequently access the blood directly or via the lymphatic system [3]. Additionally, Filoviruses are stable and can be infectious as aerosols, by the oral and conjunctival routes [4] making them a bioweapon concern. Symptoms of Ebola virus disease occur after an incubation period ranging from several days to 3 weeks [5]. Although, the virus circulates throughout the body, the highest concentrations are in the liver, kidney, spleen, and lungs. Disease onset is rapid, beginning with fever, headache, malaise, chills, and loss of appetite, muscle and joint pain. The symptomatology proceeds with abdominal pain, nausea, vomiting, coughing, sore throat, diarrhoea, bleeding, capillary leakage, maculopapular rash often developing 5 to 7 days into the illness. Fatal cases progress to coma, shock, culminating in death [6]. As the result of the rapid nature by which Ebola haemorrhagic fever progresses, death or survival of infected individuals is determined very soon after infection. Current therapeutic regimens are unable to eradicate the virus and to restore health in Eb-infected patients. Several promising candidates for vaccine are under investigation [7]. In the light of available data, there is a dire need to develop curative strategies such as immune-therapeutic approaches to address the public health burden posed by Ebola. Filoviruses primarily replicate in mononuclear phagocytes inducing production of proinflammatory cytokines [8], this process may explain the damage to the lymphatic organs. Studies aimed at understanding the biology of ZEBOV and the mechanisms of pathogenesis in humans and animal models indicate that mononuclear phagocytic cells and endothelial cells are sites of early replication and play critical roles in the pathogenesis of Ebola haemorrhagic fever [3,9,10]. In the present study by using microarray dataset analysis we detect the expression of genes that are transcriptionally activated in peripheral blood mononuclear cells (PBMCs) in response to the immune dysfunction caused by ZEBOV infection. We disclosed specific contribution and the vicious cycle of immune activation driven by macrophages (MΦ) on the disease outcome. The identification of key genomic signatures might be useful for the future development of early diagnosis of EBOLA infection. Gaining understanding of the immune responses and immunopathology induced by these viruses may be critical to the advancement of the Filoviruses vaccine platforms and to post exposure treatment strategies for Filoviruses infected individuals.
Filoviruses are enveloped, non-segmented, negative-sense RNA viruses that exhibit a unique heterogeneous filamentous structure. The viral genome encodes seven structural proteins, nucleoprotein (NP), polymerase cofactor (VP35), matrix protein (VP40), glycoprotein (GP), replication-transcription protein (VP30), minor matrix protein (VP24), and RNA-dependent RNA polymerase (L). EBOV also expresses at least one secreted non-structural glycoprotein (sGP). The first step of replication occurs through interaction between GP and some cellular molecules, followed by endocytosis and macropinocytosis [10]. The subsequent fusion of the viral envelope with the host cell endosomal membrane releases the viral proteins NP, VP35, VP30, L and RNA genome into the cytoplasm, the site of replication. During replication, full-length positive-sense copies of the viral genome are synthesized, which serve as templates for replication of negative-sense viral RNA synthesis. At the plasma membrane, NP-encapsidated full-length viral RNAs and the other viral structural proteins are assembled with VP40 and GP and incorporated into enveloped virus particles that bud from the cell-surface [11]. Filoviruses show broad tissue tropism. Their preferred target cells are monocytes/macrophages, dendritic cells, endothelial cells and hepatocytes [12]. The infection of these cells is important for haemorrhagic manifestation and immune disorders [12]. Apart from direct tissue damage, resulting from EBOV replication an explanation for the poor acquisition of ZEBOV immunity in naturally exposed populations is that the virus negatively modulates the immune system of the host, preventing the development of specific immune responses, such as the induction of proinflammatory cytokines, depletion of T lymphocytes and coagulation abnormalities. The viral surface GP appears to be crucial in controlling the tropism and pathogenesis of Filoviruses infection [13]. GP is highly glycosylated with large amounts of N- and O-linked glycans, most of which are uniformly located in the middle one-third of the GP, designated the mucin-like region (MLR) [14] signaling 1 (SOCS1) through interacting with Toll-like receptor 4 (TLR4) [15,16]. The GP affects the innate immunity by inducing proinflammatory cytokines and suppression of anti-inflammatory cytokine. Besides innate immunity, GP disturbs host adaptive immunity exerting a dual effect by its mucin domain, masking epitopes on GP that could otherwise be recognized by antibodies and shielding major histocompatibility complex I (MHC) molecules, thereby impairing antigen presentation by the host cell [16]. GP undergoes proteolytic cleavage by host proteases such as furin 15, which produces two subunits, GP1 and GP2, linked by a disulfide bond. The GP1 subunit mediates viral attachment, most likely, through the MLR [17]. The GP2 subunit catalyses fusion of the viral envelope and host cell membrane [18]. Endosomal proteolysis of EBOV GPs by cysteine proteases such as cathepsins B and L plays an important role in inducing membrane fusion [19,20]. Also, soluble glycoproteins seem to be involved in immune evasion [16]. GP causes endothelial cell activation, as indicated by upregulation of cell adhesion molecules, and decreases endothelial barrier function, potentially leading to edema and shock [17,21]. Another effect of GP on the host adaptive immune response in vitro is the presence of a 17-mer peptide that causes apoptosis of CD4+ and CD8+ cells [22]. Additionally, GP counters the host antiviral activity of tetherin Bst-2 or (CD317), which retains virus particles on the cell surface after budding, through a direct interaction of GP with tetherin [23]. Recently, the cellular receptor T-cell immunoglobulin and mucin domain 1 (TIM-1) has been identified as candidate genes involved in ZEBOV entry [24]. Ectopic TIM-1 expression in poorly permissive cells enhanced EBOV infection, as well as TIM-1 binds ZEBOV GP. However, the fact that not all cell types, which are naturally permissive for Filoviruses, express the above-mentioned molecules implies that Filoviruses may utilize multiple cellular proteins for infection of a wide variety of cells. Recent evidence suggests that endo/lysosomal cholesterol transporter protein Niemann-Pick C1 (NPC1) is essential for Filoviruses infection. The cleavage of the GP1 subunit by endosomal cathepsin removes heavily glycosylated regions to expose the putative RBR, which is a ligand for NPC1 and mediates membrane fusion by the GP2 subunit [18,19]. As well as GP, VP24 and VP35 are important determinants of pathogenicity in vivo, through their ability to counteract the IFN-induced innate immune response [20,25]. VP24 preventing IFN-α/β-induced gene expression counteracts the antiviral effects of IFN-β by interacting with karyopherin-α1 [26], which mediate nuclear import. This interaction neutralizes the innate immune response preventing nuclear accumulation of phosphorylated signal transducer and activator of transcription 1 (STAT1) [27]. To overcome the potent properties of the innate immunity, EBOLA developed other mechanisms mediated by VP35, which similarly to VP24, blocks IFN-α/β production [20]. in vitro, VP35 prevents the activation of interferon regulatory factor 3 (IRF-3) by delaying the virus-induced phosphorylation of this protein [22] through several mechanisms. First, the binding of VP35 to double-stranded (ds)RNA prevents dsRNA-mediated activation of retinoic acid-inducible gene 1 (RIG1) [22] which would normally result in phosphorylation and nuclear translocation of IRF-3 and subsequent expression of the IFN-α/β genes [22]. Second, VP35 is phosphorylated by κB kinase epsilon (IKK-ε) and TANK-binding kinase 1 (TBK-1), thereby impairing the interaction of these kinases with their substrates IRF-3 and IRF-7 thus avoiding their phosphorylation [23]. Finally, through an interaction with the protein inhibitor of activated STAT1 (PIAS1) and the ubiquitin-conjugating enzyme Ubc9, VP35 is involved in the SUMOylation of IRF-3 and IRF-7, resulting in decreased transcription of the IFN-β gene [28]. The ability of VP35 to bind dsRNA also inhibits the phosphorylation of dsRNA-regulated protein kinase R (PKR), which would otherwise result in the phosphorylation of translation initiation factor eIF-2α and, thus, blocking protein synthesis [29]. Yet, another in vitro effect of the ability of the carboxyl terminus of VP35 to bind dsRNA is the suppression of RNA silencing [29]. The amino terminus of VP35 affects maturation of dendritic cells thereby suppressing upregulation of co-stimulatory molecules and pro-inflammatory cytokines and attenuating their ability to activate CD4+T cells [29].
The innate immune system is the keystone for recognizing and eradicating Filoviruses infections. Monocytes/macrophages represent a complex and nuanced population of immune cells, which are critical effectors and regulators of the inflammatory response to diseases and infections, acting as a bridge between innate and adaptive immunity by monitoring the microenvironment through an array of surface receptors and secreting appropriate cytokines and chemokines [24,30]. Monocytes released from bone marrow into the circulation migrate into most tissues of the body, where they differentiate into mature MΦs, including liver Kupffer cells, brain microglia, lung alveolar MΦs, peritoneal MΦs, adipose tissue MΦs and bone osteoclasts, to name a few. The mainstreams of mononuclear phagocytic cells in the bloodstream are monocytes. In both primate and human models, most of the major organs, including the liver, lymph nodes, and spleen, show high titers of Ebola virus. Virus production in mononuclear phagocytic cells seems to be the major source of the high viremia in naturally and experimentally infected hosts during the critical early stages of the infection. High viremia subsequently allow the direct infection of important secondary target cells, such as endothelial cells [9,31,32]. In experimental Ebola infection of cynomolgus macaques the disseminated intravascular coagulation is, most likely, a direct result of replication of EBOV in lymphoid MΦs [33]. Early and sustained infection in monocytes, also plays a central role in the occurrence of viral haemorrhagic fever, through the expression of proinflammatory and antiviral cytokines, including interferon-alpha (IFN-α), interleukin-1 (IL-1), IL-6, IL-8, IL-12, and TNF family members ( e.g., TNF-α and TRAIL), the induction of the expression of cell adhesion molecules on endothelial cells ICAM-1, VCAM-1, E-selectin, and P-selectin) and of coagulation factors (e.g., tissue factor), leading to activation of the extrinsic coagulation pathway and ultimately to endothelial cell destruction and permeability [34-36]. In addition, the production of proinflammatory cytokines in Ebola- infected monocytes and MΦ, and of chemokines, such as IL-8 and GRO-α causes diapedesis of leukocytes [37,38]. The chemokines IL-8 and GRO-α are not only chemoattractant for leukocytes but also trigger monocytes to activate vascular endothelium, suggesting an additional role in monocyte recruitment [39]. Therefore, extravasation of activated, infected monocytes may be a mechanism by which EBOV spreads from the bloodstream into organ tissues during the second stage of infection. This could explain the pantropism associated with severe Ebola infections, and this would not necessarily depend on damage to the endothelium. The massive leukocytes mobilization strongly amplifies the inflammatory process, also the excessive cytokines production results in the shock development [40]. In fact, the supernatants of Filoviruses-infected macrophages increased endothelial permeability an effect that was mainly driven by TNF-α [41], the endothelial permeability is one of most important events during shock development [42], which is the major cause of death in Ebola haemorrhagic fever [41]. In addition, clumping of monocytes is involved in the pathogenesis of filovirus haemorrhagic fever [43]. Monocyte clumps in vessels in vivo dramatically influence the rheology of the bloodstream, mainly in small venules, leading to thrombus formation in infected patients. Intravascular clump formation could alter the coagulation pathways, a phenomenon observed in many clinical cases [44,45]. Particularly the intrinsic one, as observed in infected monkeys [46-48].
Microarray technology has the advantage of allowing simultaneous analysis of the expression of thousands of genes in a sample. The breadth and sensitivity of the analysis allows identification of pathways affected and cascades of gene expression alterations that would be impossible to achieve by other methods. We employed the reference gene expression dataset GSE8317 and GSE5099 obtained from the NCBI Gene Expression Omnibus (GEO http://www.ncbi.nlm.nih.gov/geo/) database to identify differentially expressed genes in PBMCs of ZEBOV infected cynomolgus macaques and in human monocyte-to-macrophage differentiation and polarization. Using this approach, we were able to assess differential gene expression for each gene in a comparable manner.
Many viruses take advantage of the MΦs polarization system to enhance their own growth and virulence. Focusing specifically on MΦs population highly responsive in terms of altering their polarization pattern, we evaluated the expression of genes that typically distinguish M0 and M1/M2 subsets by microarray dataset analysis. The fractions of monocyte subsets were defined by CD14, CD68 and CD33 expression varied during the first 6 days post infection (Figure 1A). The expression pattern of CD14, which represents the major monocytes subsets constituting ~90% of the circulating blood monocyte population showed the higher expression at 4th day post infection. In contrast, CD33 that represent immature monocytes was higher at 3th day post infection, remarkably decreased at 4th day post infection, when CD14 were significantly higher. In contrast, the level of CD68, which represents activated macrophages, was higher at 3th and at 5th day post infection. These results indicate that significant changes in monocyte maturation and activity occur early during ZEBOV infection. Between the mediators mainly involved in the upregulation of M1 we found that the pro-inflammatory cytokine TNFα, IL1β and IL6 resulted significantly induced at 5th day post infection, implying that M1 polarization exacerbate ZEBOV infection in sustaining inflammation (Figure 1B). The outcome of infection depends on a delicate balance between appropriate and inappropriate induction of these mediators. It has been reported that survivors of Ebola infection have 100- to 1,000-fold lower peak viremia and significantly greater virus-specific antibody responses than fatal cases, who exhibit marked immunosuppression [36,49]. Survivors of infection most likely can mount a more rapid and more effective innate immune response, which curbs virus replication sufficiently to allow time for the development of adaptive immune responses. Survivors of Ebola infection have an early and short-lived rise in serum chemokines, indicative of innate immune system induction [35,50]. Curiously, both survivors and non-survivors do not differ in their serum levels of important regulators of adaptive immunity such as IFNα, IFNγ, IL12, IL17, or TNFα [48]. M2a subsets are marked by the upregulation of several surface molecules including CD209 a lectin-surface receptor. C-type lectins are a family of Ca2+-dependent carbohydrate-recognition proteins that play crucial roles in innate immunity. CD209 gene encodes DC-specific intercellular adhesion molecule-3 grabbing non-integrin (DC-SIGN) [51] which is a type II transmembrane protein committed to antigen uptake. We observed that CD209 reached the maximum levels at 4th day post infection (Figure 1C). Its presence on MΦ depends on the tissue type and state of activation [52]. CD209 is present in certain tissue MΦ such as in the alveoli and lung. The significant increase of CD209 could in part explain ZEBOV infection in the respiratory tract, causing severe inflammation of the lung and consequently rapid and difficult breathing. In HIV infection, the binding of the viral envelope glycoprotein to DC-SIGN might induce a conformational change that enables a more efficient interaction with CD4 and/or the chemokine receptor. It is reasonable that ZEBOV utilize the same strategy used by HIV, the binding of DC-SIGN to glycoprotein may facilitate or stabilize one of these transitions. In addition, binding of viral particles to DC-SIGN may focus or concentrate them at the surface of the M2 and thus increase the probability that entry will occur after they bind to the receptor complex on target cells. Similarly, to HIV, which is captured by the same receptor [53], in vivo, DC-SIGN-mediated entry of ZEBOV in APCs both M2 and DC), conceivably, influencing virus persistence and host immunity. To counteract these inflammatory mechanisms, it appears that pathogenic ZEBOV has evolved 'silencing' strategies, involving C-type lectins [39,40]. In fact, membrane-anchored cellular C-type lectins facilitate Ebola entry [54]. The carbohydrate chains on Ebola GP are recognized also by other cellular C-type lectins such as dendritic cell- and liver/lymph node-specific ICAM-3-grabbing non-integrin DC/L-SIGN [55] and human MΦ galactose-type C-type lectin (hMGL) [41]. Therefore, MΦ, expressing C-type lectins, are thought to be the preferred target cells of Filoviruses [12,42,43]. Indeed, primary MΦ cultures transduced for C-type lectin expression greatly increased their susceptibility to virus infection [56]. Therefore, C-type lectins are the most important molecules, probably as attachment factors, directly involved in Ebola tropism at the cellular level. Accordingly, soluble mannose-binding C-type lectin is involved in protection from lethal Ebola virus infection in a mouse model [57]. Interestingly, the ability to utilize the C-type lectins (i.e., DC-SIGN and hMGL) to promote cellular entry was correlated with the different pathogenicity among Filoviruses [44,45]. The CD163 is a feature of the "alternatively activated" MΦs that contrast the classical activated M1-type MΦs [6,46]. CD163 is a scavenger receptor for the endocytosis of haemoglobin and haemoglobin/haptoglobin complexes and is nearly exclusively expressed in monocyte/macrophages. CD163 is induced by IL-10 and glucocorticoids while proinflammatory cytokines like TNF-α reduce its expression. The IL-6, which exerts pro- and anti-inflammatory effects, depending on the signaling pathway activated, strongly upregulates CD163. Anti-inflammatory cells involved in the down-modulation of inflammation express high level of CD163 that controls immune response. Ligands of the TLR2, 4 and 5 stimulate ectodomain shedding of CD163 thereby releasing soluble CD163 (sCD163) which mediates cellular uptake of free haemoglobin. In our analysis, CD163 expression showed an undulating pattern with an increment at 3th day, a decrement at 4th day to reach the maximum level at 5th day post infection, whereas at 6th day reverted to the basal levels (Figure 1D). Soluble CD163 circulates in blood, increases in serum of critically ill patients and in infectious diseases. Serum concentrations of sCD163 predict comorbidity and mortality in some diseases and are suitable biomarkers for diagnosis, prognosis and therapeutic drug monitoring in several inflammatory disorders. For example, in some virus such as African swine fever virus (ASFV) and the porcine reproductive and respiratory syndrome virus PRRSV), CD163 binding is crucial for the virus infection in different ways. The ASFV exploits CD163 for attachment and internalization [58], whereas PRRSV employs CD163 binding for virus uncoating [59]. Further investigation should establish the physiological role of CD163 and the consequences in EBOLA infection. Interestingly, the microarray analysis showed that CD302, was significantly low at 4th, 5th and 6th day. CD302 is an unconventional lectin receptor that not only plays roles in endocytosis/phagocytosis but also in cell adhesion and migration [60] for these futures could be developed as a target for therapeutic manipulation. Regarding the genes distinguishing M2b subsets significant modulation was observed for CCR2, IRF4 and CCL18 (Figure 1D). The chemokine/chemokine-receptor pairs CCL2, CCL8/CCR2 are monocyte chemotactic chemokines. They showed the maximum levels at 3th day of ZEBOV post infection. Recruitment of monocytes/macrophages in a particular organ in response to inflammation is a vital response to eliminate invading pathogens through phagocytosis. Nevertheless, the massive presence of MΦ can have also detrimental effects, as confirmed by the finding that CCR2 was highly expressed in HCV inflamed livers [61]. Remarkably, CCR2 has both proinflammatory and anti-inflammatory actions. The proinflammatory role of CCR2 is dependent on APCs and T cells, whereas the anti-inflammatory role is dependent on CCR2 expression in regulatory T cells. However, so far, there is no evidence suggesting that any one of them affects clinical disease outcome in patients with Ebola infection. CCR2-deficient mice are resistant to the induction of sensory neuropathies [62]. Discovery of drugs, which block several upregulated chemokine receptors, may be very effective if they are upstream of CCR2 expression. Therefore, any attempts to influence CCR2 expression to achieve beneficial effects should be carefully considered. The CCL18, which is a potent chemoattractant increased at 4th day, peaked at 5th day and reverted to basal levels at 6th day post infection. CCL18 is one of the most highly expressed chemokines in chronic inflammatory diseases including idiopathic pulmonary fibrosis [63], bronchial asthma [64] and atopic dermatitis [65]. In addition, CCL18, not only possesses fibrotic activity but if expressed at high level in the lung and mainly by alveolar MΦs act as chemoattractant for naive CD4+ T lymphocytes and immature monocyte-derived dendritic cells [66], which may lead to the development of tolerogenic immune response. Strikingly, IRF4 production was significantly inhibited at 4th, 5th and 6th day post infection suggesting that M2 subsets acquire a inflammatory phenotype. The IRF4 belonging to IRF transcription factor family, plays pivotal roles in the differentiation and function of T and B-lymphocytes. It is a crucial mediator of M2 polarization [67]. Many of the genes with IRF4 binding sites including mannose receptor (MR), and metalloproteinase 9 (MMP9), have been associated with the development and key functions of the M2 phenotype. IRF4 inhibits TLR signaling by interacting with MyD88 in the cytoplasm, but the molecular mechanisms of M2 induction by IRF4 remain unknown [68]. Moreover, IRF-4 could function as a repressor by binding to an IFN stimulated response element ISRE) of some genes, such as those encoding MHC class I, IFN-stimulated gene 15 (ISG15) and TRAIL [69]. In summary the microarray analysis indicated that the 3th and 4th days post infection was characterized by a massive presence of M1/MΦs (Figure 2). Unlike bacterial pathogens, which generally tend to thrive within and encourage production of M2-subset, viral pathogens more commonly activate M1 polarization. This inflammatory phenotype is often correlated with disease severity. In ZEBOV infected PBMCs the most prominent population was M1 subset during the days post-infection. Nevertheless, a significant presence of M2 subset was observed until the 5th day (Figure 2). The strong recruitment of MΦs at 4th day, and the persistence over the analysed period, indicates that MΦs are highly responsive in terms of shifting their polarization pattern during the course of ZEBOV infection. Therefore, it is conceivable that the viral survival strategy takes advantage of both M1 and M2 subsets as means to reach different goals: M1 subset as mean to recruit fresh immune cells to spread the infection, and M2 subset as a reservoir of replication. This can also be inferred from the ability of proinflammatory cytokines and chemokines from ZEBOV-infected MΦs to enhance virus replication and dissemination. This behaviour is common in viral diseases. For instance, the viral protein NS3 of Hepatitis C virus (HCV) enhances cytokines production in THP-1 MΦ [70]. Furthermore, activation of MΦ with TLR agonists triggers the secretion of TNFα, which promotes ZEBOV systemic inflammation. ZEBOV shows a more complex relationship with MΦ polarization. It is capable of polarizing monocytes towards an anti-inflammatory M2 phenotype including high expression of the scavenger receptor CD163 and suppression of MHC expression. Based on these results, we can envisage M2a-polarized MΦ as "Ebola-transmitting MΦ" in that characterized by the selective expression of DC-SIGN/CD209, which, being downregulated in M1 subset, represents a potentially useful marker for discriminating M1 and M2/MΦ.
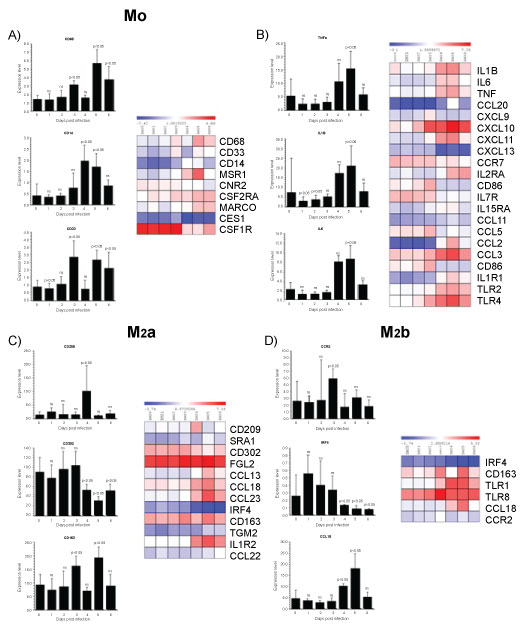 Figure 1: Gene enrichment analysis.
Figure 1: Gene enrichment analysis.
Gene expression dataset GSE5099 was obtained from the NCBI Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) database. GSE5099 included human monocyte-to-macrophage differentiation and polarization transcriptional profiling data. For this dataset, macrophages were obtained by culturing monocytes for 7 days in complete RPMI 1640 medium supplemented with 100 ng/ml M-CSF, while macrophage polarization was obtained by stimulating cells with 100 ng/ml LPS plus 20 ng/ml IFN-γ for M1 polarization or 20 ng/ml IL-4 for M2 polarization. Monocytes at day 0 of culture were used as reference. We calculated the False Discovery Rate to generate a list of up-regulated and down-regulated genes. We used a threshold of p < 0.001 and fold change > 2. The same statistical approach was used to calculate the significantly modulated genes in GSE8317. The list of significantly up and downregulated genes for each comparison was considered for further analysis. Gene enrichment analysis was performed using the Chi-square test with Yates' correction. Comparison groups were represented by macrophage, M1 and M2 from GSE5099 and day 1 through 6 as compared to the pre-infection baseline from GSE8317. A p value < 0.05 was considered to be statistically significant.
View Figure 1
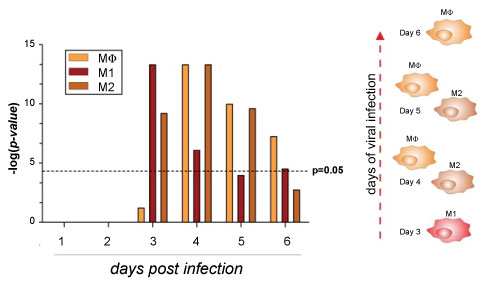 Figure 2: Macrophages population in ZEBOV infected PBMCs.
Figure 2: Macrophages population in ZEBOV infected PBMCs.
Gene enrichment analysis was performed using the Chi-square test with Yates' correction. Comparison groups were represented by macrophages, M1 and M2 subsets from GSE5099 from day 0 to day 6 as compared to the pre-infection baseline from GSE8317. P value < 0.05 was considered to be statistically significant.
View Figure 2
Monocytes-macrophages express pattern recognition receptors (PRRs) to recognize molecular pathogen associated molecular patterns (PAMPs) [71]. The TLRs are PRRs that participate in the first line of defence against invading pathogens recognizing distinct PAMPs [48]. TLRs are type 1 membrane spanning receptors consisting of an extracellular leucine-rich repeat (LRR) domain, a transmembrane- spanning domain and a cytoplasmic Toll-IL1 receptor-resistance (TIR) domain. TLRs encompass molecules located at the cell surface and others located into the intracellular endosomal compartment. Most of the TLRs family are involved in responses to viral infection [72]. The microarray dataset analysis indicated that in infected PBMCs, ZEBOV triggers host responses through TLRs 1, 2 and 4. In particular, TLR 4 induction stated from the 2th day post infection and reached the higher levels at 5th day. TLR1 and TLR2 induction started at 3th day and reached the maximum levels at 4th day post infection. Interestingly, the expression of TLR-1, 2 and 3 decreased at 6th day post infection (Figure 3A), suggesting that TLRs 1,2 and 3 provides protective immunity ZEBOV virus. Immune responses against a number of viruses are, at least partially, dependent upon TLR2 [73,74] and occur across many different cell types. For example, Epstein-Barr virus (EBV) in monocytes [75] and herpes simplex virus (HSV) in microglial cells [76] all elicit TLR2-dependent responses. Additionally, TLR2- Respiratory syncytial virus (RSV) interaction is important not only in cytokine release to restrain viral replication, but also in neutrophil migration and dendritic cell activation in the lung [77]. Conversely, in some cases of viral infection TLR2 may be helpful to the virus benefit. In a report of patients with chronic HCV infection, it was demonstrated that the HCV core protein engaging TLR2 induced the expression of IL-10 and TNF-α cytokines from monocytes, through a reduction in IFN-α release from plasmacytoid DCs (pDCs) and pDC apoptosis [78]. Moreover, in patients with chronic hepatitis B infection, TLR4 is overexpressed on monocytes and modulates the activities of regulated T cells (Treg) which may contribute to immunotolerance [79]. As well, the recognition of Ebola virus glycoprotein by TLR4 leads to the production of proinflammatory cytokines and thus is thought to mediate viral immunopathogenesis contributing to detrimental immunity. The outcome of a virus-TLR interaction is complex and depends upon the particular TLR and the virus in question as well as the host species. It is now appreciated that immune responses Ebola infection requires the contribution of TLR1, 2 and 4. Further studies revealing how TLRs signals can mediate harmful immune responses following Ebola infection, should aid to develop drug targets of TLR signalling pathways (i.e. viral inhibitors of TLR signalling) in order to improve Ebola infection. Therapeutic manipulation of TLR-agonists with anti-viral activities could be employed either as treatments for viral infection or as vaccine adjuvants.
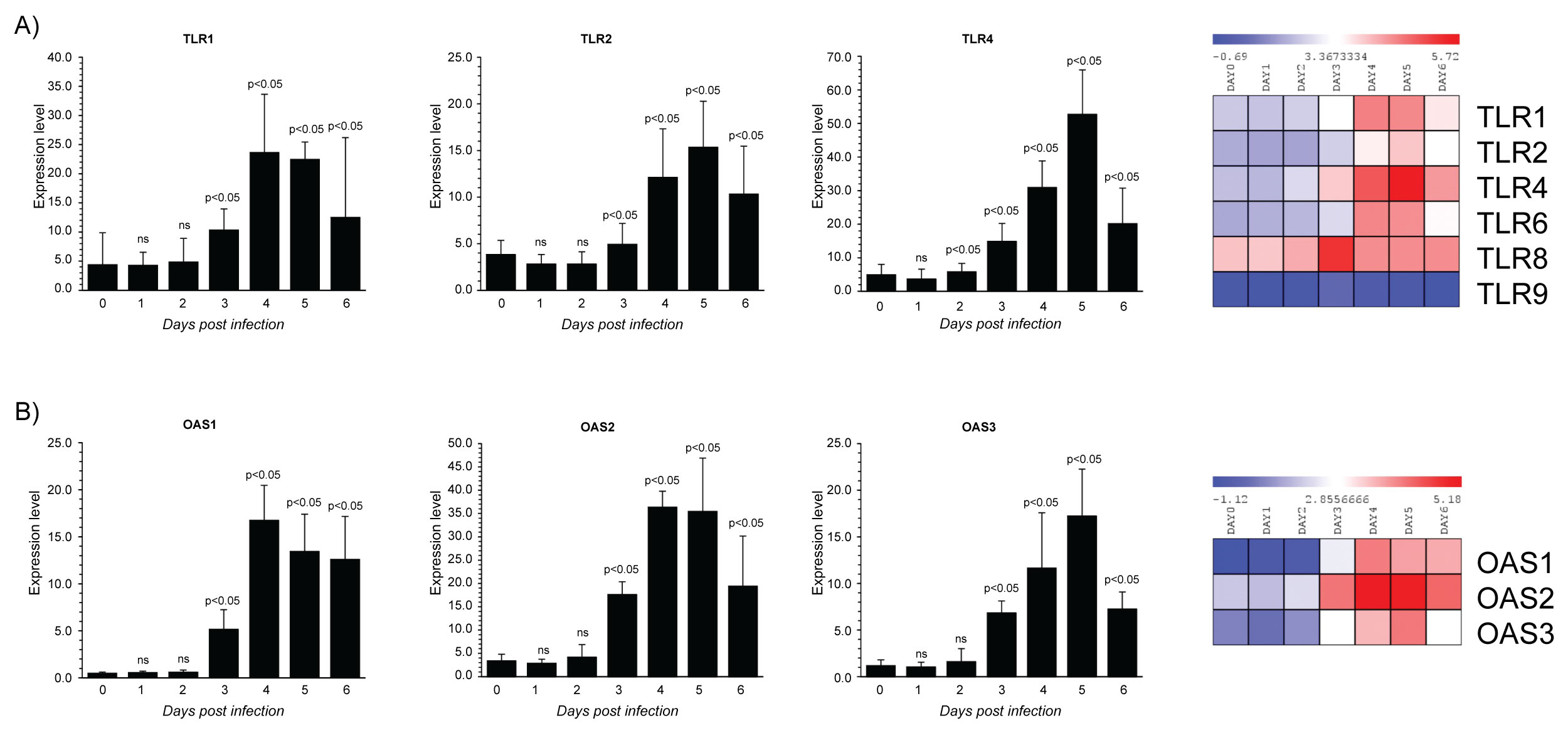 Figure 3: Gene expression dataset GSE5099 of TLRs and OASs family members.
Figure 3: Gene expression dataset GSE5099 of TLRs and OASs family members.
A) TLRs expression in Zebov infected PBMCs; B) OASs expression in Zebov infected PBMCs. P value < 0.05 was considered to be statistically significant.
View Figure 3
The IFN-inducible antiviral state is mediated, in part, by the 2',5,'-Oligoadenylate [2-5] synthetase (OAS)/RNaseL system. 2',5'-oligoadenylate synthetases OASs) belong to the nucleotidyl transferase family. They generate 2',5'-oligoadenylates from ATP in response to viral double-stranded RNA, which binds and activate a latent RNAse known as RNAseL. RNaseL restricts viral infections by degrading viral and cellular RNA, inducing autophagy and apoptosis, and producing RNA degradation products that amplify production of type I IFNs through RIG-I-like receptors. Thus, OAS and RNAseL are the major mediators of dsRNA degradation in virus-infected cells. RNA degradation products are recognized by RIG-I helicase and MDA5 - the major cytoplasmic sensors of virus infection in mammalian cells [80]. There are four OAS enzymes that are produced in various splice forms: OAS1, OAS2, OAS3, and OASL [81], which are localized in different compartments within virus infected cells [11]. The OAS enzymes are potent antiviral this is because they control the effects of virus infection [82]. In fact, genotypes of OAS have been reported to correlate with outcomes of HCV infection [83], liver fibrosis in chronic HCV infected patients [84] and are linked to patient IFN-responsiveness during anti-HCV therapy [85]. Our analysis indicated that the levels of expression in OAS genes were differentially regulated during virus infection, probably reflecting differential transcriptional control and/or distinct functions [85]. In particular, little changes in OAS 1, 2 and 3 expression at day 0, 1 and 2 post infection were observed, whereas starting from the 3th day post infection OAS1, 2 and 3 transcripts were significantly upregulated (Figure 3B). The major induction of both OAS1 and OAS2 expression was observed at the 4th day post infection. Regarding OAS3, the maximum level of expression was reached at 5th day post infection, and similarly to OAS2, at 6th day post-infection the expression decreased reaching the same levels observed at 3th day. This data indicates that OASs are ineffective for mediating interferon's antiviral effect against ZEBOV. Although the OAS1 gene is recognised as an IFN-response gene and IFN-α and IFN-β induce several antiviral OAS genes OAS2 [82] in the case of HCV infection was reported that elevated baseline viral loads and of OAS were correlated with IFN non-responsiveness [86]. Further, was reported that high basal levels of OAS/RNaseL in MΦs reduce, rather than increase, virus induction of IFN-β. RNA damage and apoptosis caused by RNase L are likely reasons for the decreased IFN-β production in virus-infected MΦs [87]. Therefore, in ZEBOV infection it is conceivable that as in the case HCV infection OASs overexpression fails against viral loads. This result warrant further investigations to clarify failure to IFN-based therapies.
The human leukocyte antigen (HLA) system, the MHC in humans, has long been considered the most important region in the human genome with respect to infection and inflammation [88]. In humans, HLA complex consists of more than 200 genes located close together on chromosome 6. Genes in this complex are categorized into three basic groups: class I (HLA-A, -B, and -C), class II (HLA-DR, -DQ, and -DP), and class III. Interactions among HLA-restricted T lymphocytes, B lymphocytes, natural killer (NK) cells, and cytokines influence immune response to viral infection. HLA class I and II molecules are expressed as cell surface antigens that bind to peptide epitopes on CD8+ T cells and CD4+ T cells, respectively. Effective presentation of viral antigens by the HLA system induces good immune response. The HLAs are good targets for viruses to escape from immune-surveillance. While the downregulation of classical HLA molecules by viruses have been often described, the upregulation of non-classical immune-tolerant molecules is a new area of research. Evaluating the microarray dataset analysis in PBMCs of infected cynomolgus macaques, we found that between HLA class I molecules, HLA-E were upregulated at 5th day post-infection, HLA-F increased at 4th day, reached the higher levels at 5th day and at 6th day post-infection reverted to the level observed at 4th day (Figure 4A). Regarding HLA-G, the most significant enhancement was observed at 3th day and at 5th day post-infection. Suggesting that HLA-E, HLA-F and HLF-G are upregulated to be recognized by inhibitory receptors, thus inducing the down-regulation of anti-viral response. Indeed, certain viruses, including HIV-1 and human cytomegalovirus, can take advantage of this HLA-E-mediated pathway to evade killing by NK cells [89]. HLA-G is important to control the host immune response during infection [90]. Additionally, it was demonstrated that HSV type 1 and rabies virus as well as HIV type 1 induce HLA-G expression in neurons and T cells, respectively [91]. HLA-G is a non-classical HLA-class I molecule showing immunotolerance properties [92]. HLA-G inhibits the lytic activity of both decidual and peripheral blood NK cells as well as Ag-specific CTL and allogeneic proliferative responses [93,94]. Additionally, HLA-G expression is induced following IAV infection in alveolar epithelial cells, the first targets of IAV [95]. The evidence indicating that HLA-G inhibits the immune system and the function of T lymphocytes specifically directed against influenza [96], and that influenza escapes from immunosurveillance by the upregulation of HLA-G, suggest that ZEBOV embraces the same behaviour. HLA-G is expressed during pregnancy [96] and is mainly expressed at the maternal-foetal interface where it was identified as a key molecule in maternal-foetal tolerance [97]. Therefore, this data may explain the major susceptibility of pregnant women to Ebola [97]. The role of HLA-G during virus infections could have a clinical relevance as it was demonstrated that HLA-G is induced after several virus infections such as in HCMV and HIV infected patients [98,99]. Ultimately, HLA-G expression could paralyse the immune system before Ebola infection. Knowing more about HLA-G may thus represent a crucial step towards the development of new HLA-G based therapeutic against viruses. HLA class II molecules constitutively expressed by APCs, present peptidic antigens to CD4+ T cells and in this manner are essential to adaptive immune responses against pathogens. The HLA-II genes are distributed over three highly polymorphic contiguous genetic loci-HLA-DR, HLA-DP, and HLA-DQ which encode conventional histocompatibility molecules. Interestingly, in our examination between HLA class II molecules, the expression of HLA-DPA1, HLA-DRA and HLA- DQA2/ HLA-DQA1 resulted downregulated indicating an anergic state of infected PBMCs (Figure 4B). HLA class II molecules and the Ag-processing pathway may be targets of ZEBOV for escaping CD4+ T lymphocyte immunosurveillance. The expression of MHC-II molecules in the cells can also be induced by IFN-γ through the induced transcription of Class II transactivator (CIITA), CIITA appears to be a vulnerable target for viral modulation of MHC-II gene expression because is it masters the regulation of the expression of MHC-II elements, including HLA-DR, HLA-DQ, HLA-DP, HLA-DM, HLA-DO, and invariant chain/CD74. CIITA enhances the transcription of MHC-II genes through interaction with transcription regulatory proteins and forms a stable complex through binding to the regulatory module of MHC-II promoters [100]. In this context, HCMV infection of U373 MG cells reduced the level of IFN-γ-induced CIITA transcripts by more than 10-fold, with a concordant inhibition of induced MHC-II DR synthesis [101]. Several reports have described analogous immune evasion strategies adopted by other virus, as revealed by studies on both murine and human CMV showing that the virus blocks IFN-γ-inducible MHC class II transcription by inhibiting Jak1 expression and disrupting IFN-γ-stimulated signaling pathway in murine [102], and interfering with MHC class II transcription and CIITA activation in human [103]. Similarly, varicella-zoster virus infection of human foreskin fibroblasts can prevent IFN-γ-induced CIITA expression and expression of MHC-II molecules [104]. Other strategies for the down-modulation MHC class II surface expression by HCMV are promoting the proteasome-mediated degradation of DR-α and DM-α molecules by glycoprotein US2 [105]. Only recently analogous immune evasion strategies have been described for EBV. EBV GP42 protein binds to HLA class II molecules at their various stages of maturation and impairs TCR mediated activation of Ag-specific Th cells in an HLA class II-dependent manner [106]. A truncated soluble EBV GP42 protein, generated by proteolytic cleavage of full-length gp42 in the endoplasmic reticulum during the EBV lytic infection, can mediate Ag-specific evasion from Th cell recognition by blocking the TCR-HLA class II-peptide association [107]. A study reported that EBV lytic infection is associated with the reduction of MHC class II expression [108]. The data reported in this study confirm that HLA antigens may be involved in several steps of the immune deficiency of Ebola infected subjects and thus contribute to the pathophysiology the disease. Reduced levels of some HLAII molecules could play a role in the progression of the disease and affect both the clearance of Ebola virus and complement mediated antibody responses. The mechanisms underlying modulation of HLA expression in ZEBOV infection needs deeply investigation.
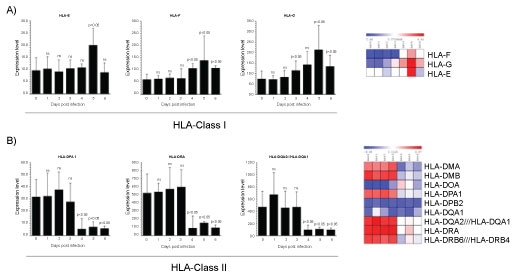 Figure 4: Gene expression dataset GSE5099 of HLA I and HLA II.
Figure 4: Gene expression dataset GSE5099 of HLA I and HLA II.
A) HLA class (I E, F,G) in Zebov infected PBMCs; B) HLA class II (DP,DQ, DR) in Zebov infected PBMCs. P value < 0.05 was considered to be statistically significant.
View Figure 4
Mammalian chitinases are lysosomal enzymes belonging to the family of glycoside hydrolase-18, located on the chromosome adjacent to the MHC paralogue on genes implying a crucial role for chitinases in innate and adaptive immunity. Mammalian chitinases are divided into chitinases and chitinase-like proteins depending on their enzymatic activity. The chitinase-like proteins or chitinase- like-lectins (Chi-lectins) include chitinase 3-like 2 (YKL39) and chitinase 3-like 1 (YKL40), and chitinase domain containing 1 (CHID1) among others [109]. For most of the mammalian chitinases important biological roles in chronic inflammatory diseases have been identified [110-113]. They may function in the recognition of PAMPs encoded in chitin, thereby signaling to the host immune system to mount an appropriate pathogen directed attack. Our analysis showed a different modulation of Chitinase-like-lectin (CHI3L1, CHI3L2 and CHID1). CHI3L1 was significantly upregulated at the 5th day reaching the maximum levels at 6th day. Interestingly, this was the only Chitinase showing the higher expression at 6th day post-infection (Figure 5A). CHI3L1 is a 40 kDa glycoprotein, due to the substitution of an essential glutamic acid residue to leucine, entirely lacks glycohydrolase enzymatic activity. Nevertheless, still retains a functional chitin-binding motif acting as chi-lectin through a preserved hydrophobic substrate binding cleft [114]. Biological activities of CHI3L1 embrace regulation of cell proliferation, adhesion, migration and activation. CHI3L1 is not synthesized under state of health, but, several experimental evidences suggest that CHI3L1 plays an important role in the processes of inflammation and tissue remodelling [113,115]. It has been reported that CHI3L1 stimulation significantly activates NF-κB transcription in human colon cell lines and enhances the production of TNFα, IL-8, RANTES and eotaxin [116]. These findings suggest that CHI3L1 may play a crucial role in the pathogenesis of colitis by enhancing the production of proinflammatory cytokines and chemokines. Additionally, CHI3L1 is a vascular cell adhesion and migration factor that may have a role in processes leading to vascular occlusion and heart development [117]. CHI3L1 is differentially upregulated in cirrhotic liver on the end-stage of hepatitis C virus HCV) induced liver cirrhosis [118]. Increased production of CHI3L1 is indication for liver pathology [118] and can be a useful non-invasive marker for evaluation of the degree of fibrosis as well as efficiency of therapy in patients with HCV-associated liver disorders [118]. Overall, this evidence indicates that elevation of CHI3L1 levels might exacerbate the inflammation. By facilitating the production of potent chemical mediators, the migration of phagocytic cells or some others non-specific factors could increase the systemic changes. CHI3L2 increased at 3th day and was significantly upregulated at the 5th day (Figure 5B). CHI3L2 or YKL-39 lack of any chitinase activity but retain putative lectin properties [119], thus CHI3L2 acts as a pseudo-chitinase with retention of chitinase-like ligand-binding properties [119]. CHI3L2 was originally identified as an abundantly secreted protein in primary culture of human articular chondrocytes [120] and displays anabolic properties in cartilage. Recently, it was demonstrated that CHI3L2 mRNA was expressed on a very low level in MF differentiated in the presence of IFN-γ or IL-4 but increases significantly in M1 subset, whereas in M 2 reverted to base levels [121]. Moreover, a combination of IL-4 and TGF-β in MΦs strongly stimulated the expression of CHI3L2 [122]. Nevertheless, IFN-γ has inhibitory effect on the production of CHI3L2 in MFs [121]. Therefore, also CHI3L2 enhancement may be influenced by the altered cytokine production. The CHI3L2 promoter sequences showing two STAT1 and two NF-κB binding sites suggests that CHI3L2 could play a crucial role as an inducer of immune responses related to EBOLA. Further investigations are indispensable to pinpoint the biological activity of CHI3L2 in viral EBOLA infection. Conversely, the expression of CHID1 showed a significant decrement at 4th and 6th day post-infection (Figure 5C). CHID1 or stabilin-interacting chitinase-like proteins (SI-CLP) is the most recent identified human Glyco_18 domain-containing protein [122]. It was found as an interacting partner and sorting ligand for the multifunctional receptor stabilin-1, which is specifically expressed on subpopulations of tissue macrophages and sinusoidal endothelial cells in liver, spleen, lymph node and bone marrow [122,123]. In parallel with stabilin-1, expression of SI-CLP mRNA was found strongly upregulated in macrophages by IL-4. CHID-1 is the only chitinase-like protein, which is upregulated by glucocorticoids [123]. Stabilin-1 is one of the major scavenger receptors expressed by liver sinusoidal endothelial cells (LSEC) and guarantees proper clearance of the peripheral blood from potentially noxious agents and thus contribute to tissue homeostasis throughout the whole body. The deficiency of stabilins results in multiple pathologies that may be caused by a circulating factor or factors that evade clearance by LSECs. Therefore, it is conceivable that CHID-1 acts like stabilin-1 contributing to tissue homeostasis not only in the liver, but also in other organs. The inhibition of CHID-1 by ZEBOV infection could contribute to the impairment of its clearance and, so, could be another mechanism that may enhances virus dissemination.
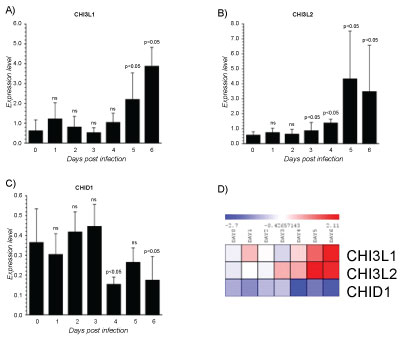 Figure 5: Gene expression dataset GSE5099 of Chitinase-like-lectins (CHI3L1, CHI3L2 and CHID1).
Figure 5: Gene expression dataset GSE5099 of Chitinase-like-lectins (CHI3L1, CHI3L2 and CHID1).
A) CHI3L1 expression in Zebov infected PBMCs; B) CHI3L2 expression in Zebov infected PBMCs; C) CHID1 expression in Zebov infected PBMCs. P value < 0.05 was considered to be statistically significant.
View Figure 5
The data collected in this study spanned from as early as the first day of fever onset to acute period, providing an exceptional opportunity to investigate the whole spectrum of host responses during the disease process. The samples from earliest time points were particularly valuable in understanding the differences in pathology between monocyte/macrophages at early stages of ZEBOV infection. Put together, these data indicate that MΦs play a critical role in the ZEBOV disease severity and provided some new biological insights. We observed a gradual evolution in the gene expression patterns over time from day 0 to day 6, usually with a more significant change at 4th day, resulting in the separation of the early acute (day 0 to day 3) and late acute phases (day 3 to day 6). The early acute and late acute phases were represented by two waves of functionally distinct gene clusters (Figure 6). Detecting the gene expression patterns appears evident that although hosts have adapted use of the MΦs strategies to galvanize multiple additional layers of immune defence, EBOLA have evolved multiple tricks to avoid the immediate antiviral effects of MΦs and uses this cellular system as a Trojan horse. To the best of our knowledge, this study was the first study to present a systemic analysis of the full dynamics of the host response in ZEBOV infected PBMCs and provided solid data to highlight the importance of the timing in the collection of the clinical samples. Strengthening the role of macrophage's arsenal in the battle against EBOLA could advance the understanding of the ZEBOV-mediated disease progression, which will provide enormous support for future clinical research, diagnostics and vaccine development.
 Figure 6: Graphical representation showing the main regulated genes in the different days of infection in monocyte/macrophages subsets.
View Figure 6
Figure 6: Graphical representation showing the main regulated genes in the different days of infection in monocyte/macrophages subsets.
View Figure 6
Source of Gene expression dataset GSE5099 was obtained from the NCBI Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) database.
We declare that we do not have any financial interest or personal relationships with other persons or organizations that could influence our work.