We report a case of a two-year-old girl presenting with progressive multiple bony swellings over the neck and back, diagnosed as Fibrodysplasia ossificans progressiva (FOP) based on radiological and genetic tests. FOP is an extremely rare disorder of heterotopic ossification which leads to progressive immobility. Efforts to correct deformities by surgical resection further stimulate new bone formation and lead to clinical worsening. Classically, FOP presents as malformation of the great toes and progressive skeletogenesis. It can be misdiagnosed in early stages, and flare-ups usually occur during the first decade of life, resulting in increasing disability. Currently preventive measures and symptomatic management of flare-ups is the commonly used approach. Drug trials are underway for development of more definite treatment of this rare condition.
Heterotopic Ossification, Fibrodysplasia, Genetic Disease, Exoskeleton
Fibrodysplasia ossificans progressiva (FOP) is an extremely rare genetic disorder with an estimated prevalence of 1 per 2 million worldwide [1], often mislabelled as osteochondromas. FOP also known as Munchmeyer’s disease or Stoneman disease is characterized by soft tissue masses progressing to heterotopic ossification (HO) in the subcutaneous tissue or muscles, resulting in progressive immobility and disability. Great toe malformation is a classical finding in these children. Conductive hearing loss due to middle ear ossification or sensorineural hearing loss due to involvement of the inner ear, cochlea, and auditory nerve can also occur [2]. Atypical features of FOP are categorized as FOP- plus and FOP variants [3]. It is caused by a sporadic mutation in the ACVR1 gene, resulting in dysregulated bone morphogenetic protein (BMP) signalling [4]. This case report illustrates the characteristic radiographic findings of the disease. Prompt recognition and diagnosis can prevent unnecessary surgical interventions and ensure the implementation of essential precautions, such as avoiding intramuscular injections in young children, which can promote local tissue ossification.
A two-year-old female child, a resident of Central Asia, who was the fourth and youngest child of a nonconsanguineous marriage; presented with complaints of progressive multiple, hard, painless swellings over the back since the age of 4 months. It started from the occiput then involved the neck, upper chest, shoulder, back, and left thigh. This resulted in neck stiffness with restricted neck and spinal mobility which was progressive. There were no systemic features like fever, weight loss, failure to thrive, hepatic or renal disease, and no features suggestive of an autoimmune disease, or an underlying malignancy. There were no prenatal, perinatal, or postnatal complications or similar complaints in siblings or other family members. On examination, there were multiple hard bony swellings over the posterior aspect of the neck associated with a restricted range of motion, anterior chest wall, upper back, and lateral aspect of the left thigh (Figure 1). There was no lymphadenopathy, hepatosplenomegaly, or bony tenderness. The systemic examination including respiratory and cardiovascular was also normal. Routine blood investigations were unremarkable with no evidence of hypercalcemia (serum calcium- 10.4 mg/dL serum phosphate- 5.1 mg/dL, serum alkaline phosphate- 331 U/L). Anteroposterior and axial radiographic evaluation revealed contiguous soft tissue ossification along the paravertebral soft tissue extending throughout the spine and involving the anterior chest wall, and the lateral aspect of the left thigh (Figure 2). The CT scan showed sheet-like calcification over the anterior chest wall, upper back, and bilateral paraspinal locations from the occiput to the lumbar region, suggestive of calcification universalis (Figure 3). Similar calcification was seen in the left thigh likely secondary to childhood vaccinations. X-ray of both feet revealed bilateral shortened first metacarpals consistent with clinical diagnosis of FOP (Figure 2A). Diagnosis was confirmed by whole exome sequencing (WES) showing a mutation in ACVR1 gene, with a heterozygous pathogenic variant at (c.617G > A) (p.Arg206His) at exon 6 (ACFMG class 1). The patient was offered supportive therapy with two weeks of tapering corticosteroids for flare-ups. The parents were counselled about the natural course of the disease including prognosis and advised to protect from trauma and avoid surgical interventions.
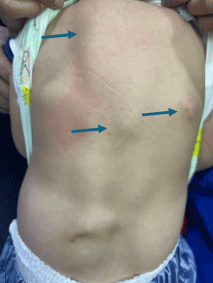 Figure 1: Multiple irregular, non-tender, bony hard swellings (green arrows) on the back:
View Figure 1
Figure 1: Multiple irregular, non-tender, bony hard swellings (green arrows) on the back:
View Figure 1
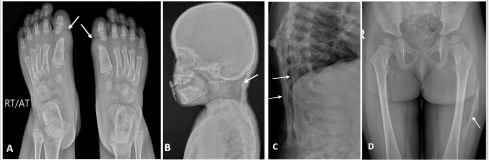 Figure 2: (A) Plain radiograph (anteroposterior view) of bilateral feet showing great toe malformation due to shortened 1st metatarsals. (B, C) Plain radiographs of neck and thoracodorsal spine (lateral view) showing heterotopic bone formation of posterior neck muscles and back. (D) Plain radiograph of the pelvis and bilateral thigh (anteroposterior view) showing ossification in the lateral aspect of the left thigh.
View Figure 2
Figure 2: (A) Plain radiograph (anteroposterior view) of bilateral feet showing great toe malformation due to shortened 1st metatarsals. (B, C) Plain radiographs of neck and thoracodorsal spine (lateral view) showing heterotopic bone formation of posterior neck muscles and back. (D) Plain radiograph of the pelvis and bilateral thigh (anteroposterior view) showing ossification in the lateral aspect of the left thigh.
View Figure 2
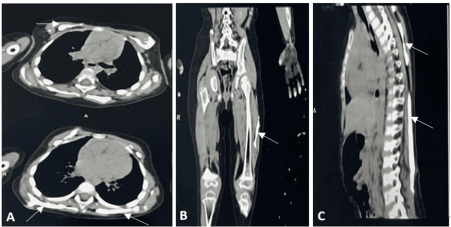 Figure 3: Computed tomography revealed extensive heterotopic ossification (white arrows) in (A) anterior chest wall; (B) left thigh and (C) back.
View Figure 3
Figure 3: Computed tomography revealed extensive heterotopic ossification (white arrows) in (A) anterior chest wall; (B) left thigh and (C) back.
View Figure 3
FOP is an extremely rare genetic disease characterized by skeletal malformations and soft tissue masses that progress to heterotopic ossification. This was first described by John Freke in 1740 as “progressive ossifying fibrous dysplasia”. FOP is not known to be affected by race, ethnicity, gender, or geographic distribution [1]. The disease entails a lifetime of painful metamorphosis and progressive immobility and disability. Two classical clinical features FOP include malformation of the great toes; and progressive HO in specific spatial patterns.
Most cases arise from sporadic mutations, but some are genetically transmitted (autosomal dominant). The primary pathology is dysregulation of the BMP signaling pathway [5], most caused by ACVR1 gene mutation (c.617G > A; R206H). This is a single heterozygous missense mutation in the glycine-serine (GS) activation domain located on chromosome 2q23-24 [4]. The gene encodes for activin receptor IA (ACVR1)/ activin like kinase 2 (ALK2) which is a BMP type I receptor. Patients with this mutation show the classical phenotype. Other associated mutations include L196P, R258S, P197/F198lL, R202I, Q207E, G325A, G328W/G328E/G328R, G356D and R375 [6]. To date, all detected ACVR1 mutations are gain-of-function mutations leading to increased BMP signalling [7, 8].
Great toe deformities include hallux valgus, abnormal segmentation, pseudo epiphysis, and first metatarsal fusion [7, 9]. Typically, during the first decade of life, sporadic episodes of painful soft tissue swellings, described as “flare-ups” [10] occur, which leads to musculoskeletal disability. Most patients are wheelchair-bound by the end of the second decade of life. HO follows an axial to appendicular, cranial to caudal, and proximal to distal distribution [11]. Neck stiffness is an early finding and can precede the appearance of HO. Other skeletal features include clinodactyly, short malformed thumbs, short broad femoral necks, and proximal medial tibial osteochondromas [3, 12, 13]. Chest wall involvement and spinal deformities including kyphosis, thoracic lordosis, and scoliosis, can lead to thoracic insufficiency syndrome (TIS) [7, 14]. Life-threatening complications include progressive immobility, jaw ankylosis leading to severe weight loss, pneumonia, and right-sided heart failure due to TIS. Certain muscles like cardiac and smooth muscle, diaphragm, tongue, and extra-ocular muscles are spared in FOP.
In our case report, radiological imaging revealed HO highly suggestive of FOP which was further confirmed by WES. Currently, there is no definitive treatment for FOP. Bracing is ineffective for spinal malformations and surgical attempts to remove ossifications lead to new bone formation. Supportive medical management includes treating flares with steroids and preventive measures like avoiding trauma are important. Muscle relaxants can be used for muscle spasms. Non-steroidal anti-inflammatory medications, COX-2 inhibitors, leukotriene inhibitors, mast cell stabilizers, and amino bisphosphonates are used for chronic discomfort and ongoing flare-ups [15].
Shaikh et al [16] have discussed the following potential treatment strategies including inhibition of ACVR1/ALK2 gene-related pathway and osteoblastic progenitor cell activity. Palovarotene, a selective retinoic acid receptor ɣ agonist has been FDA approved for heterotopic ossification in females > 8 years and males > 10 years [17]. Genetic therapies using CRISPR-Cas9, RNA interference (RNAi), and Adeno-associated Virus (AAV) vectors are also underway. Activin A antibody (Garetosmab), ALK2 inhibitor (Saracatinib), inhibition of Glycogen synthase kinase-3 (GSK-3) β, and targeting peroxisome proliferator-activated receptor (PPAR) γ are being explored.
We report a typical case of FOP with radiographic findings corresponding to ossifications of various soft tissues subsequently confirmed by genetic testing. FOP is an ultra-rare disease that can be missed at initial presentation and inappropriately treated by surgical excisions of lesions. An early accurate diagnosis may help alleviate the suffering and preserve body function. Ongoing clinical trials targeting molecular pathway may provide a potential curative treatment for FOP.
NIL
NIL for all authors
NIL
The manuscript has been developed as per institutional ethical policies. Written informed consent was taken from the mother for use of patient details.
Mridul Singh and Anushka Aggarwal contributed equally.