Background: Numerous patients suffering from orphan diseases still lack a treatment. Pharmaceutical companies play a crucial role in the advancement of orphan drug development. This systematic literature review aims to identify and categorise current incentivising factors for pharmaceutical companies to develop orphan drugs.
Methods: EMBASE and MEDLINE databases were systematically searched for terms related to orphan drug development incentives for pharmaceutical companies. Research findings were qualitatively evaluated and categorised into themes.
Results: 752 publications were found in the initial search. Full-text review was performed on 64 publications and 14 publications were included in the final systematic review. Incentivising factors were split into four categories: Regulatory factors in the USA, regulatory factors in the EU, clinical trials and patient voice.
Conclusion: Patient voice is a crucial factor in engaging and incentivising pharmaceutical companies in orphan drug development. Numerous policies are currently in place across both USA and EU targeting different stages of orphan drug research. Further qualitative data is required to evaluate stakeholder views on policy effectiveness.
Orphan disease, Orphan incentives, Orphan medicinal product, Pharmaceutical, Rare disease
ODA: Orphan Drug Act; COMP: Committee for Orphan Medicinal Products; EMA: European Medicines Agency; HUD: Humanitarian Use Device; NME: New Molecular Entity; PRIME: Priority Medicines: OMP: Orphan Medicinal Product; InSPiRe: Innovative Methodology for Small Populations Research; IDeAl: Integrated Design and Analysis of Small Populations Group Trials; Asterix: Advances in Small Trials Design for Regulatory Innovation and Excellence; FDA: Food and Drug Administration; EURORDIS: European Organisation for Rare Diseases: NORD: National Organisation for Rare Disorders
The Orphan Drug Act (ODA) passed in the USA in 1983 was the first of its kind to address the need for development of rare disease drugs. The Act intended to provide financial incentives for drug developers who would historically overlook rare disease research and development due to its limited profitability. Almost two decades after the US adopted the ODA, the EU passed Regulation No 141/2000, or the Orphan Drug Regulation, in 1999. Similar to the ODA, it aimed to encourage industry to develop drugs for rare diseases. The regulation introduced the procedure for orphan designation in Europe and the incentives orphan medicines would receive. It also established the Committee for Orphan Medicinal Products (COMP), a committee under the European Medicines Agency (EMA) responsible for evaluations of orphan designation applications and general protocol assistance [1].
These regulations have received both praise and criticism over the years. Whilst the 2233 designations granted between 2000-2019 targeting a range of treatment areas [2] can be viewed as a success, 95% of rare diseases still lack approved treatments [3]. Therefore, incentives remain crucial for the development of orphan drugs by pharmaceutical companies.
The objective of the review was to identify key themes in the literature regarding incentives for pharmaceutical companies to develop orphan drugs.
Two electronic databases were searched: EMBASE and MEDLINE (both accessed through Ovid).
Search terms were generated from key words in the title and those identified through an informal review of the literature (Figure 1).
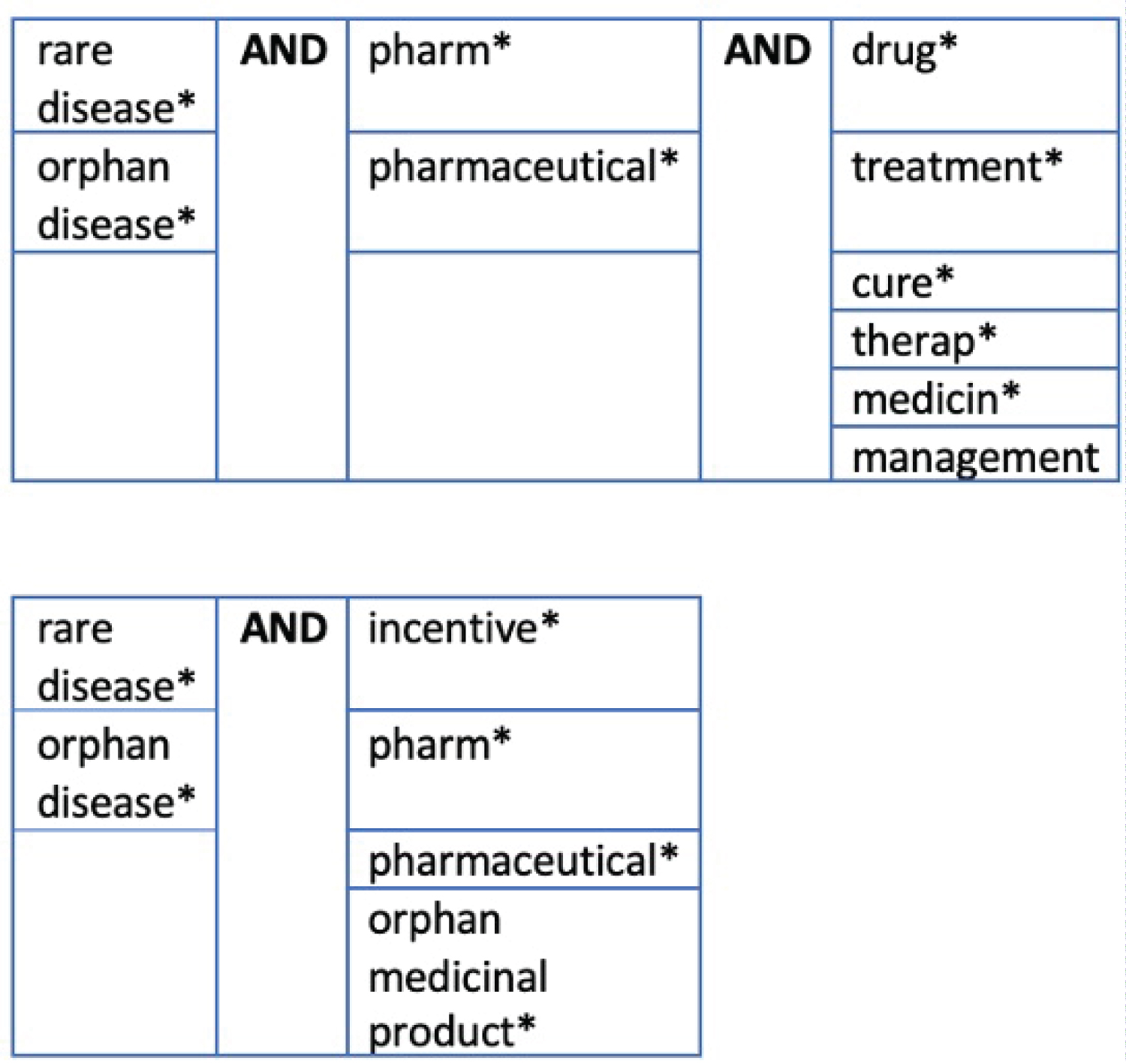 Figure 1: SLR search terms. View Figure 1
Figure 1: SLR search terms. View Figure 1
Inclusion criteria were papers from the previous 5 years (2015-2020), based in either Europe or the USA and any study design. Non-English papers, those not available in full text, and papers focussing on cost and disease-specific clinical papers were excluded.
Two databases, EMBASE and MEDLINE, were used as our search terms required an advanced search function which was not available on other databases. Therefore, to maintain consistency within the search process these two databases were used. Additionally, these databases were most suitable for the research aim and provided adequate topic coverage. The 5-year time period was deemed appropriate to avoid out-dated papers and to provide an understanding of the current rare disease landscape. Papers focusing on practices in both Europe and USA were included as both regions have arguably played a crucial role in influencing orphan drug development. To thoroughly explore incentivising factors and obtain comprehensive qualitative data, all study design types were considered.
Articles retrieved were first reduplicated and screened by title and abstract. Full text screening was then performed against defined inclusion and exclusion criteria (Figure 2). Papers were screened independently by two reviewers.
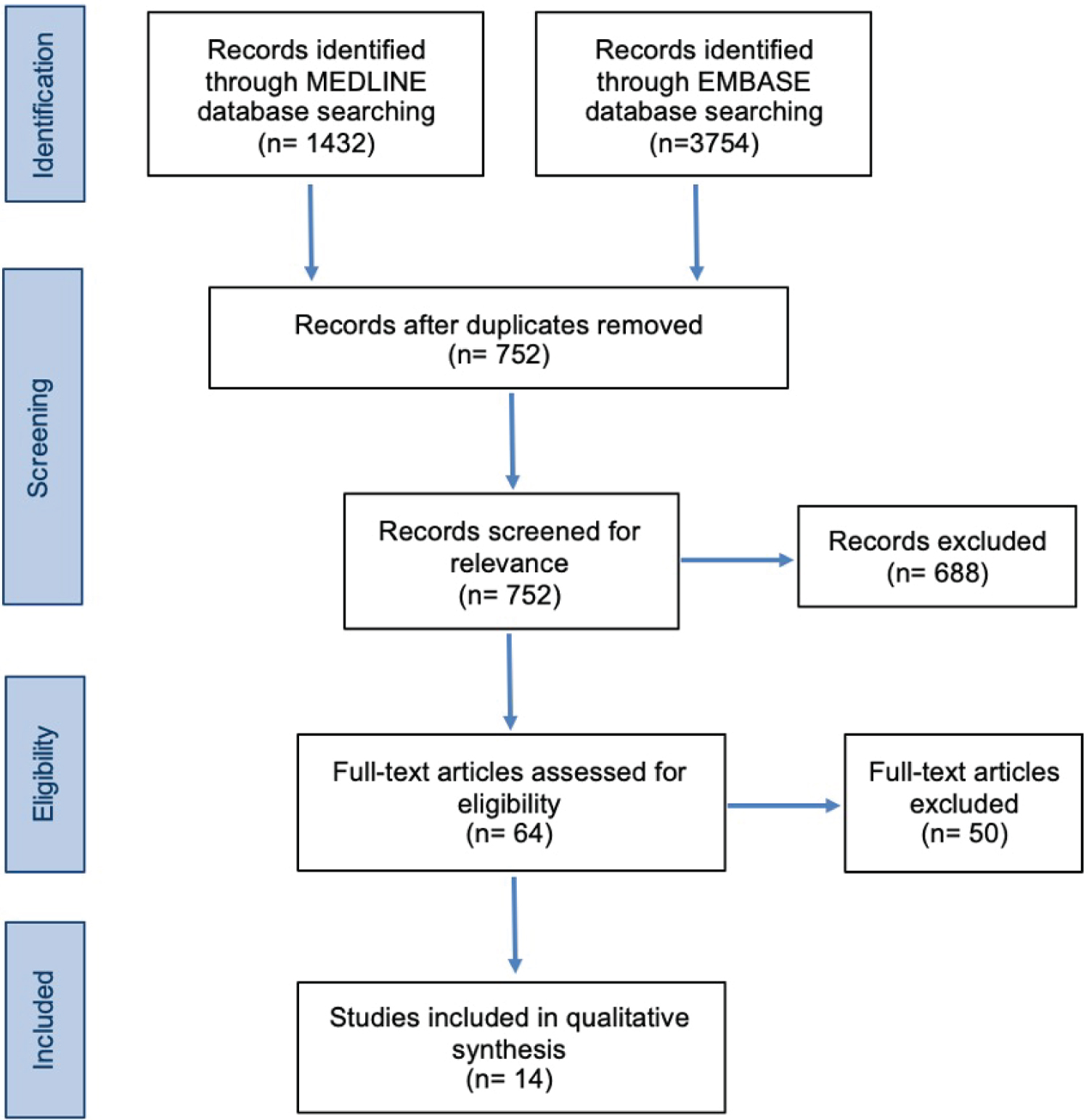 Figure 2: SLR Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) Diagram. View Figure 2
Figure 2: SLR Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) Diagram. View Figure 2
Articles were carefully evaluated and assessed based on appropriateness to the main research objective, potential for bias and quality of reporting.
14 studies were eligible for inclusion from the databases used. Papers were then categorised into key themes which were identified as significant factors that incentivise the pharmaceutical industry to develop drugs for rare diseases (Figure 3).
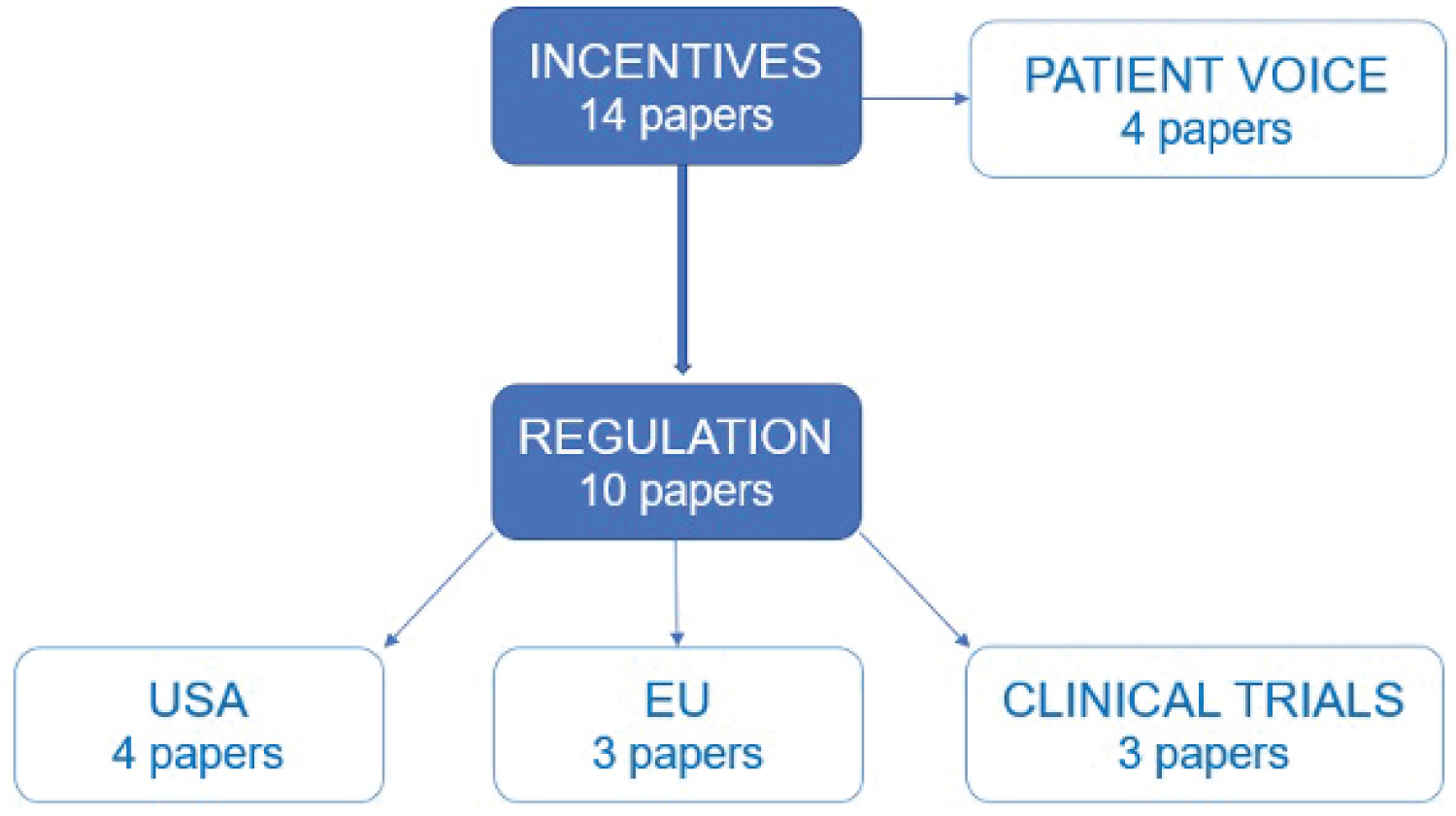 Figure 3: SLR themes. View Figure 3
Figure 3: SLR themes. View Figure 3
The findings of the literature review are presented in the following order: Regulation in the USA, regulation in the EU, clinical trials and the patient voice.
The 7-year market exclusivity, whereby another sponsor will not be approved for the same drug for the same indication during the 7 years, is considered to be one of the most important regulatory incentives driving orphan drug development, ensuring profitability for the exclusivity holder without threat of competition [4]. However, a different paper offers some critique on this policy, arguing that as new drugs are often protected by other patents which last longer than the exclusivity period; the role of market exclusivity is diminishing [5]. This was supported by the results of a retrospective cohort study carried out in the same paper, concluding that exclusivity only outlasted patents for one third of new orphan-designated drugs in the period 1985-2014. Whilst limitations of the paper must be acknowledged, such as only using publicly available data which may not be fully representative of different drug types such as biologics, the paper raises an important point about caution when using the exclusivity policy as a model for other pharmaceutical incentive programs.
Patel & Needleman [6] discusses alternative programs incentivising the development of treatments for rare diseases. The Humanitarian Use Device (HUD) Designation Program provides a separate pathway for medical devices that treat or diagnose a condition affecting no more than 8000 individuals annually in the USA. Additionally, the Rare Paediatric Disease Designation Program is specifically tailored to treatments for rare paediatric diseases affecting individuals from 0-18 years. The study reports that 164 out of 216 requests have been granted through this program as of December 2018. This program is undeniably important as the majority of rare diseases affect children.
Furthermore [7], highlights several expedited programs which have been created by the FDA to help facilitate a faster drug approval process and therefore incentivise pharmaceutical companies. Although not necessarily specific to rare diseases, the programs are applicable to any drug which can fill an unmet medical need, which is often the case in rare diseases. In brief, Priority Review reduces the time taken for FDA to review applications from 12 to 8 months for New Molecular Entity (NME) drugs and from 10 to 6 months for non-NMEs. Accelerated Approval allows approvals to be granted on a conditional basis for clinically meaningful drugs treating a serious condition. Fast Track designation enables frequent communication with the FDA to help advance the development of novel drugs for serious or life-threatening conditions. Similarly, Breakthrough Therapy provides the drug company with intensive FDA guidance throughout the development process. The paper demonstrates that there are in fact, at least at a high level, a number of policy incentives already in place in the USA which attempt to speed up the approval process and ultimately, deliver much-needed drugs to rare disease patients quicker. Indeed, many of the orphan drugs in use today have benefitted from these programs.
Similar to the expedited programs in the USA, the EMA have implemented a Priority Medicines (PRIME) program providing 'enhanced and early dialogue with EMA' to support the development process, as well as an 'accelerated assessment' program to reduce the assessment period from 210 to 150 days [8]. Furthermore, the exploratory review discusses 'exceptional circumstances', which can grant market authorisation where further evidence for a drug is unlikely to be provided due to the disease being rare, incomplete expertise or where a clinical trial would be deemed unethical. Orphan Medicinal Products (OMPs) are the most likely drugs to gain 'exceptional circumstances' approval due to a small patient population. Pharmaceutical companies can use this to their advantage as orphan drugs may have an easier regulatory route to market.
Orphan drugs were also found to have significantly lower development-to-launch times when compared to non-orphan drugs. Additionally, they are more likely to gain regulatory approval with a 93% chance of success compared to 88% for non-orphan drugs [9]. The review also highlights that in Europe, orphan drugs have a higher chance of gaining approval if the pharmaceutical company has past experience in the manufacturing of OMPs or if they have engaged with the relevant regulatory body at an early developmental stage. This reinforces the importance of transparent communication throughout the drug development process.
Gammie, et al. [10] discusses the importance of pre-licensing in incentivising pharmaceutical companies to develop drugs for rare diseases. It enables the importation of orphan drugs, which are available in other countries, into a country in which the drug is not authorised. Pre-licensing may be granted where patients with a severe or life-threatening disease lack any alternative treatment options, which is common for patients with rare diseases. Countries that permit pre-licensing encourage pharmaceutical companies to continue researching into the drug to prove clinical benefit, which may ultimately lead to authorisation. However, it must be noted that not all EU countries allow pre-licensing and where it is granted-patients may not be reimbursed by public health insurance. The EU is the sole entity with a centralised method spanning different countries for orphan drug designation and market approval. Once a pharmaceutical company has established authorisation for an orphan drug within the EU accessing other countries in the region, through initially pre-licensing, can be a favourable route to market.
Phase-1 trials are the "First-In-Man" trials and are performed at an early stage in the development of a novel medicinal product. A review found that within the initial 15 years of the EU Orphan Drug Regulation, many orphan drug applications did not require phase 1 trials as the medicinal products had been tested previously and had a recorded pharmacological profile [11]. This can be considered an incentive for pharmaceutical companies as the first stage of clinical trials has already been carried out. Furthermore, EMA authorised orphan drugs can have their indications extended to another rare disease. The possibility of using the same medicinal product for a variety of rare diseases is advantageous to the pharmaceutical industry as a single drug can be applicable to a larger patient population without further use of R&D resources. However, another paper critiques this as an incentive and highlights that designing therapies after phase 1, in the later stages of clinical trials, is associated with high failure rates which could delay drug development [12]. From the pharmaceutical industry's point of view, skipping phase 1 clinical trials may seem beneficial as fewer of their resources are required to develop a drug. On the other hand, carrying out initial trials and testing may bring additional benefits through identifying and understanding the underlying mechanisms of drug action.
The EU Framework Programme 7 (FP7), the 7th Framework Programme for Research and Technological Development created to support and encourage research in the EU, called for further research into rare diseases and led to projects such as the Innovative Methodology for Small Populations Research (InSPiRe), the Integrated Design and Analysis of Small Populations Group Trials (IDeAI) and Advances in Small Trials Design for Regulatory Innovation and Excellence (Asterix) which provided support in the design of clinical trials for small patient populations [13]. These projects enabled the pooling of expertise and discussion of current research taking place. Additionally, the authors have provided incentives into rare disease analysis through the production of free statistical software to encourage innovative research. Despite the successes of InSPiRe, IDeAI and Asterix these projects were only short-term and therefore were limited in the extent of benefit they can bring to the field. Ongoing effort and innovation are needed in this area to ensure clinical trials designs continue to be optimised for small patient populations.
Four included papers emphasised the importance of patient involvement in the process of drug development and how proactive patient advocates can help incentivise pharmaceutical and regulatory decision-making. Dang, et al. [14] highlights the current institutionalized involvement of patient organizations in several programs with the NICE, EMA and FDA. For example, COMP enables patient advocates to be 'permanent and full members with equal voting rights'. The review also observes that there have been increasingly frequent discussions of potential collaborations between rare disease patient groups and pharmaceutical companies in order to help progress research and development of treatments for rare diseases.
Bouwman, et al. [15] introduces European Organisation for Rare Diseases (EURORDIS) and National Organisation for Rare Disorders) NORD, the biggest patient organizations in Europe and USA respectively. The review also discusses the important role of patient organizations. As well as raising general awareness of rare diseases and providing support to affected families, patient organization activities include maintaining patient registries and biobank specimen collections which could be vital in providing important data for drug developers that would be difficult to access otherwise.
However, even when drug developers do reach out to patient advocates, this often happens too late in the process [16]. The commentary argues that patient engagement should happen simultaneously alongside the clinical development process. Furthermore, an important point raised in the paper is the need for developers to convert information obtained from patient groups into data that is usable for regulatory authorities.
Speid [17] supports this view that patient input is vital and examines various aspects of implementation that are often neglected in discussions. The paper argues that in order to engage patient groups, they must first be educated about the complex drug development process and its regulatory aspects through the provision of accessible information. Equipping patients with this knowledge helps them fully contribute to discussions with regulatory authorities and pharmaceutical companies. The paper also recognises the huge risk of 'exploitation of vulnerable patients' during communications between industry and patient groups. Biased presentation of information can mislead patients about the progress of a potential new drug. Therefore, the paper proposes that all patient engagement is carried out by trained professionals, all information given is accurate and unbiased and to ensure that sufficient safeguards are put in place.
The literature suggests that industry is becoming increasingly accepting of involving patients in discussions. However, the extent to which pharmaceutical companies act upon this patient feedback remains uncertain. Furthermore, engagement of patient organizations should be handled carefully to avoid any misunderstandings that may cause detriment to patients.
The scope of the review was narrowed to Europe and the USA only. Within these two regions, only the main regulatory policies that were discussed in the identified literature were included and therefore may not be fully comprehensive. The inclusion of papers from other regions would have given a broader perspective of the global rare diseases landscape.
Despite performing a thorough analysis of the regulations within the USA and EU, policies and recommendations are constantly changing within the field. It is possible that further guidance was issued after conducting the review and therefore some information may not be up to date.
The review identified key themes in the literature regarding incentives for pharmaceutical companies to develop drugs for rare diseases within the USA and EU.
Firstly, an important incentive is regulation. There are numerous policies already in place across both the USA and Europe with slightly varying practices between the two regions. The policies target different stages from expediting approvals to assisting with drug marketing, aiding orphan drug development throughout the entire process and offering benefits which non-rare disease drugs may not have. Furthermore, policies such as providing marketing authorisation in 'exceptional circumstances' demonstrate regulatory authorities' understanding of the specific challenges faced in rare diseases and how the usual requirements of non-orphan drug development, such as comprehensive data, often cannot be fulfilled. Despite receiving criticism, the regulations provide a promising starting point to encourage pharmaceutical companies to develop products to help rare disease patients.
The review also highlighted the value of engaging patient organisations in the development of treatments for orphan diseases. Collaboration between patient groups and the pharmaceutical industry appears to be taking place, although some papers argue that this occurs too late in the process and emphasise the risks of misleading interactions. Improved multistakeholder collaboration is therefore crucial for future rare disease treatment development.
We would like to thank Imperial College Business School for enabling us to complete this project as part of our degree.
Imperial College Research Ethics Committee - Science Engineering Technology Research Ethics Committee.
The contribution represents original work that has not been previously published or simultaneously submitted for publication elsewhere.
This manuscript has been read and approved by all the authors. All authors contributed equally.
The authors report no conflict of interest.
No funding was received for this research.