Fibrous dysplasia is an uncommon, bone development abnormality characterized by the replacement of normal cancellous bone by fibrous tissue and immature woven bone. Classified according to the number of affected bones, fibrous dysplasia can affect any bones of the skeletal system, with long bones the most commonly documented site of involvement. Typically, diagnosis is based on clinical, radiological and histological findings. However, due to its broad clinical spectrum, it remains a therapeutic challenge with the efficacy of various treatment approaches still highly debated. We present a case of a polyostotic form of fibrous dysplasia in a 36-year-old Indian gentleman.
The term fibrous dysplasia, first coined by Lichenstein in 1938, describes a rare, non-malignant condition in which normal cancellous bone is replaced by fibrous tissue and immature woven bone [1]. Accounting for 7% of benign bone tumours, fibrous dysplasia is caused by somatic activating mutations of the gene GNAS in a subunit of the stimulatory G-protein. This indolent, slow-growing bone disorder typically manifests in childhood or early adult life, usually with pain, bony deformity or pathological fractures. Common sites of skeletal involvement are the long bones, ribs, pelvis and craniofacial bones. Grossly, fibrous dysplasia is classified into three types - monostotic (single bone site; 70-80% of patients), polyostotic (multiple bone sites; 20-30% of patients), and polyostotic with endocrinopathies (McCune-Albright syndrome), the latter being associated with hyperpigmentation and endocrinological disorders [2]. The broad clinical spectrum of fibrous dysplasia in combination with its rarity renders its clinical recognition difficult, with diagnosis routinely based on physical, radiological and histological examination [3]. The various management strategies of this condition currently include, observation, medical and surgical treatment [1]. In this case report we focus on a case of polyostotic fibrous dysplasia in a 36-year-old gentleman with clinical and radiological features but no cutaneous abnormalities.
A 36-year-old gentleman was referred to our department following an incidental finding of an elevated alkaline phosphatase secondary to investigation for bone pain and vitamin-D deficiency. He initially presented with a one-year history of dull pain on the anterior surface of his left knee radiating to the tibia, with no associated stiffness or swelling. There was no history of local infection or trauma to the knee. He denied any other joint pain, previous fractures, headaches, fevers/night sweats, visual disturbances, hearing symptoms or sinus pressure. There was no history of endocrine dysfunction including precocious puberty and familial history for bone metabolic disorders was negative. No cutaneous abnormality was visible and clinical examination was unremarkable, with full range of motion of all joints. Blood tests showed a raised alkaline phosphatase of 1874 IU/L (RR 20-140), normal corrected calcium of 2.23 mg/dL (RR 2.20-2.60), with normal thyroid function tests and normal GGT (suggestive of bone origin for his raised ALP). His vitamin D was 13 nmol/L (RR > 50) and hence was started on and advised to continue Cholecalciferol replacement. His endocrine investigations and testicular USS were normal. Skeletal scintigraphy (Figure 1) evidenced areas of marked uptake in his skull, left scapula and left anterior tibia. Fusion SPECT CT imaging (Figure 2) showed changes suggestive of polyostotic fibrous dysplasia - intense activity in the skull with bony expansion of mixed lytic and sclerotic density, mild sclerosis and bony expansion in the left scapular spine and abnormal uptake in the anterior tibia bilaterally. Given the active disease in his skull and his tibial pain the patient was commenced on intravenous infusions of Pamidronate. Although no side effects were reported, unfortunately his pain did not respond well to Pamidronate. He was referred to neurosurgery due to the skull involvement evident on imaging, but due to the lack of neurological symptoms he was not a candidate for neurosurgery. The patient has made the decision to withhold from any further treatment at present and he remains under 6-monthly review.
 Figure 1: Technetium bone scintigram. Extensive uptake throughout the skull, marked metabolic uptake in the left shoulder and generally increased uptake throughout the axial skeleton.
View Figure 1
Figure 1: Technetium bone scintigram. Extensive uptake throughout the skull, marked metabolic uptake in the left shoulder and generally increased uptake throughout the axial skeleton.
View Figure 1
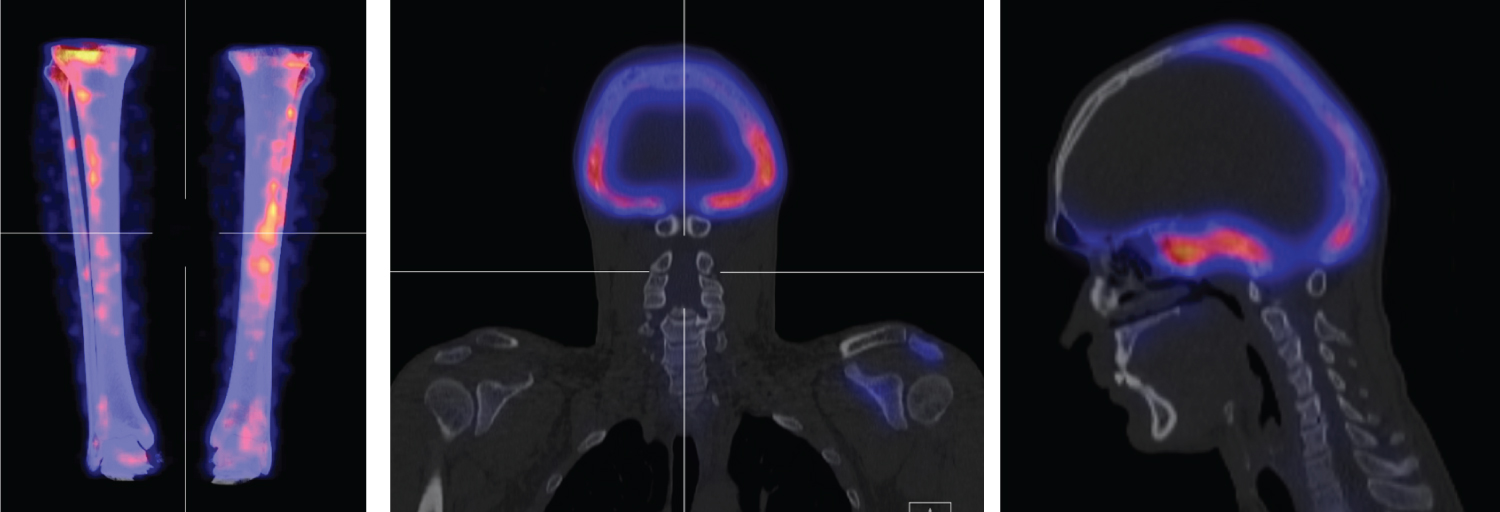 Figure 2: Fusion SPECT CT images. Intense activity in the skull with bony expansion of mixed lytic and sclerotic density, predominantly involving the occipital bone, temporal bone and sphenoid. Mild sclerosis and bony expansion evident in the left scapular spine and acromion. Abnormal uptake in the anterior tibia bilaterally, with prominence for the left side.
View Figure 2
Figure 2: Fusion SPECT CT images. Intense activity in the skull with bony expansion of mixed lytic and sclerotic density, predominantly involving the occipital bone, temporal bone and sphenoid. Mild sclerosis and bony expansion evident in the left scapular spine and acromion. Abnormal uptake in the anterior tibia bilaterally, with prominence for the left side.
View Figure 2
Fibrous dysplasia, first described by Lichenstein in 1938, describes a rare non-malignant condition affecting bone maturation [1]. Accounting for 7% of benign bone tumours [2], fibrous dysplasia describes the process where fibrous tissue and immature woven bone replaces normal cancellous bone [1].
Grossly, fibrous dysplasia is classified into three types - monostotic (single bone site; 70-80% of patients), polyostotic (multiple bone sites; 20-30% of patients), and polyostotic with endocrinopathies (McCune-Albright syndrome), the latter being associated with hyperpigmentation and endocrinological disorders [2]. With a tendency to develop pre-adolescence, this bone disorder typically manifests prior to the third decade of life [4]. However, as our case demonstrates, the age of presentation can be variable.
Commonly the initial symptom is pain in the involved limb, similar to our case. Patients may also present following a pathological fracture or be asymptomatic. Common sites of involvement are the femur, tibia, pelvis, ribs, skull, facial bones, upper extremities, lumbar spine, clavicle and cervical spine, in decreasing order of frequency [4].
Initial evaluation of a suspected case should involve laboratory studies. Elevated bone formation markers, including serum total alkaline phosphatase (with normal LFTS and GGT) or raised bone alkaline phosphatase in ALP isoenzyme testing are suggestive of active disease. Therefore, they serve as useful markers when monitoring treatment progress. In order to rule out the associated endocrinopathies, levels of testosterone, estradiol and estrone are suggested [3].
Ultimately, fibrous dysplasia can only be truly defined by clinical, radiographical and histopathological findings. Current gold standard for the diagnosis of fibrous dysplasia is a histologically-proven fibro-osseous lesion with poorly defined margins which are confirmed by radiographic findings [1]. Classical radiographic signs associated with this disease process include a "ground glass" appearance and "shpeherd's-crook deformity" which describes markedly increased curvature of the femoral neck and proximal shaft of the femur [5].
Extreme variability in the clinical presentation of this disease means patient-specific treatment is a necessity. Despite the deficit in clear guidelines for treating this condition, successful use of bisphosphonates, which inhibit osteoblastic activity, have been reported in the literature [2,5]. In an attempt to treat our patient, bisphosphonates were trialled, but then stopped due to inefficacy. This creates a therapeutic challenge in the management of this rare bone disorder.
An alternative option in management of fibrous dysplasia is surgery, including fracture management and preventative orthopaedic procedures (e.g. bone graft and curettage) [6]. However, recent literature has suggested limiting the use of surgery in polyostotic form [2].
The prevalence of malignant transformation of fibrous dysplasia is 0.4-4% [7]. Hence, periodic follow-up is suggested to observe for quiescence, regression or progression of the disease, with appropriate safety netting advice offered to patients.
Although, currently limited in treatment options, there remains hope for alternative therapy options in the near future. Tocilizumab has been suggested as a therapeutic potential in bone pain refractory to bisphosphonates [8], but more research is necessary to determine its true effectiveness. With current interest in tissue engineering techniques and the use of bone marrow stromal cells, very soon these approaches may also be potential options.
When asked to describe fibrous dysplasia in a few words, I would say "a burden". As a previously completely fit and well person, I never thought that by the end of my third decade of life, I would dread having to get out of bed just because of the intense pain fibrous dysplasia causes. I feel drained by simple daily tasks and playing cricket for my local club is an aspiration I have had to let go of. Although, I can face the pain and take some solace from the relief voltarol gel brings me, it is the uncertainty of the future which plays on my mind. From the day I received my diagnosis, every night I go to sleep worrying whether I'll still be walking tomorrow.