Pulmonary Artery Agenesis (PAA) is a rare congenital condition in which one lung has an exclusively systemic irroration and is caused by the lack of development of the 6th ipsilateral aortic arch. The affected lung is often perfused by collateral circulation developed from systemic arteries, most often from bronchial arteries [1]. The first case was described in 1868 and prevalence is estimated in 1/200,000 people. Symptoms are extremely variable as recurrent pulmonary infections, reduced exercise tolerance and hemoptysis and some individuals may remain asymptomatic for many years [2]. We report 2 cases of patients with PAA.
A 21-year-old woman presented for a history of exertional dyspnoea and atypical chest pain. No history of allergies or tuberculosis. A chest X-ray showed asymmetry of the chest walls from a reduced expansion of the right lung caused by parenchymal loss and right deviation of the mediastinum and ipsilateral elevation of the diaphragm. Transthoracic echocardiogram showed high value of estimated systolic pulmonary pressure (sPAP 45 mmHg) with normal right ventricle (Figure 1A). Right pulmonary artery not viewable. Pulmonary scintigraphy releaved low ventilation with no perfusion of the right lung, with a normal left lung (Figure 1B). Spirometric test showed a restrictive-like dysfunctional pattern with mild reduction of DLCO, 6MWT: reduction in distance walked without desaturation. CPET: normal exercise tolerance with augmented heart rate response, VO2/HR 9.7 mL/beat; VE/VCO2 45; VO2max 31 ml/kg/min, compatible with pulmonary hypertension. Angio-CT scans of the chest showed right PAA with hypertrophic ipsilateral diaphragmatic and bronchial arteries; right lung hypoplasia with diffuse bronchiectasis area (Figure 2). A right heart catheterization confirmed a borderline precapillary pulmonary hypertension without vaso reactivity and right PAA (BOPAH; sPAP 40 mmHg, mPAP 22 mmHg, PVR 1.8 UW) (Figure 3). No specific therapy for PAH was started.
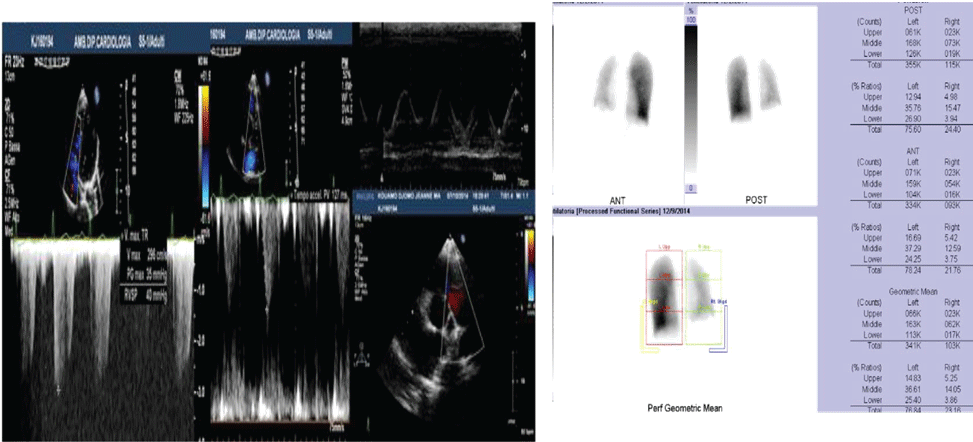 Figure 1: A) Case 1 echocardiogram showing high value of estimated systolic pulmonary pressure with sPAP 45 mmHg; B) Case 1 pulmonary scintigraphy showing low ventilation and no perfusion of the right lung.
View Figure 1
Figure 1: A) Case 1 echocardiogram showing high value of estimated systolic pulmonary pressure with sPAP 45 mmHg; B) Case 1 pulmonary scintigraphy showing low ventilation and no perfusion of the right lung.
View Figure 1
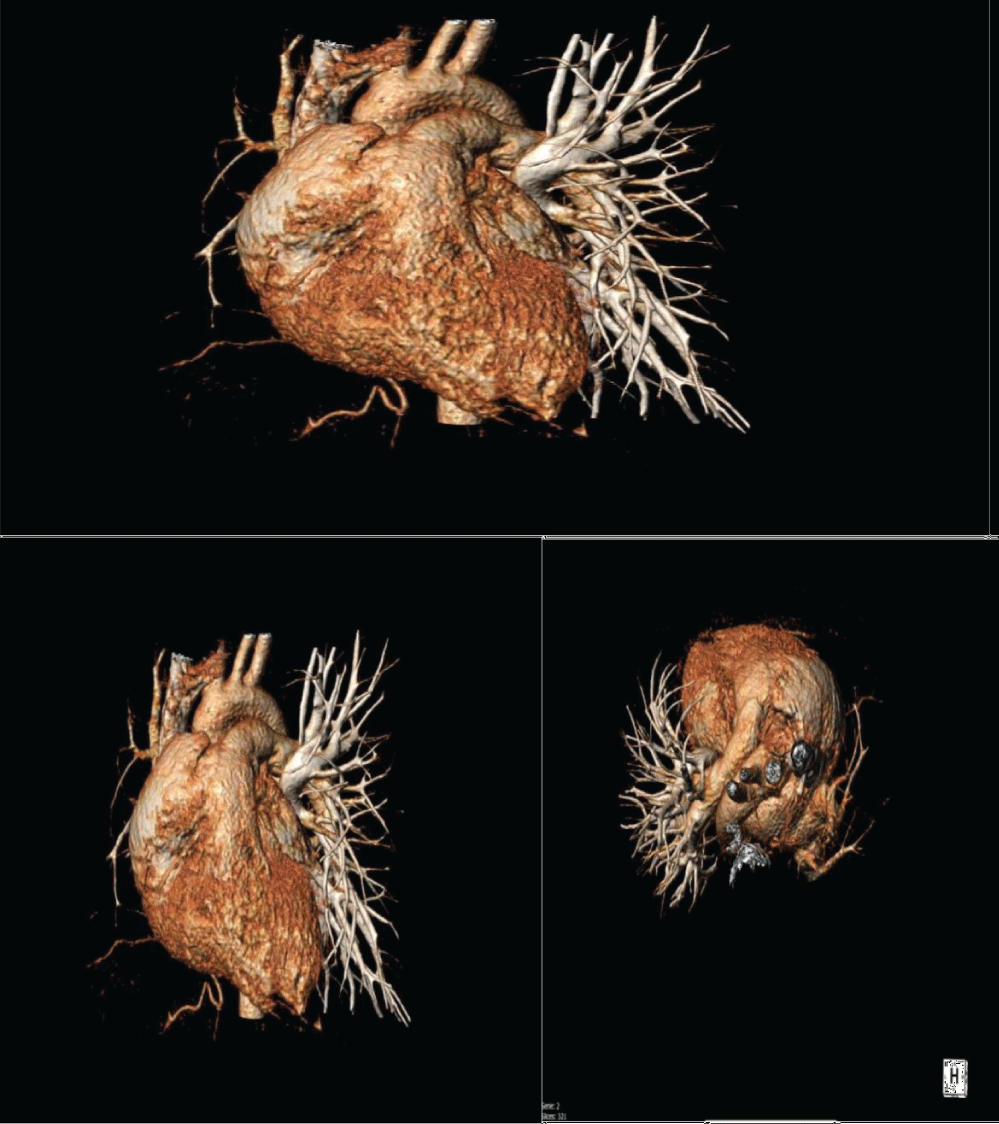 Figure 2: Case 1 CT scan 3D reconstruction showing right pulmonary artery agenesis.
View Figure 2
Figure 2: Case 1 CT scan 3D reconstruction showing right pulmonary artery agenesis.
View Figure 2
 Figure 3: Case 1 right heart catheterization and pulmonary angiography showing borderline PAH and the absence of the right pulmonary artery.
View Figure 3
Figure 3: Case 1 right heart catheterization and pulmonary angiography showing borderline PAH and the absence of the right pulmonary artery.
View Figure 3
A 42-year-old man who presented for fever, cough and worsening dyspnoea with blood oxygen desaturation (SpO2 77%) with signs of alkalosis. History of "pulmonary malformation" with small right lung.
The chest X-ray scan showed left lung pneumonia. The echocardiogram confirmed the presence of a single branch originating from the main pulmonary trunk with a sPAP 60 mmHg; Actpo 80 msec. Microbiologic culture test and procalcitonin resulted negative. The patient was discharged with an indication to perform spirometric test with DLCO and pulmonary scintigraphy. Spirometry documented the normality of forced expiratory flows, lung volumes, the flow resistances and the DLCO (KCO) 98.6%. Cardio-pulmonary exercise test showed a VO2max of 35 ml/kg/min, VO2/HR 15 (ml/beat), VE/VCO2 37, VE/VO2 40 (Figure 4). The echocardiogram performed three months after, with stabilization of the bronchopneumonic framework, documented borderline values of sPAP (36 mmHg) and pulmonary outflow acceleration time (Actpo: 123 msec) (Figure 5A). The angio-CT and hrCT scans showed parenchymal consolidation of the left lung affecting all lobes. A diagnosis of right PAA with ipsilateral pulmonary hypoplasia and main pulmonary artery ectasia was done; hypertrophic systemic bronchial and intercostal collaterals (Figure 5B); Right heart catheterization showed normal values of pulmonary pressures (mPAP 15) and pulmonary vascular resistance (PVR 1.6). Some genetic disorders play an important role in lung morphogenesis, including mutations of BMPR2 and TTF-1 factor (thyroid transcription factor-1). Molecular analysis of the first case showed the presence mutations of BMPR2 as compound heterozygosity: Exon 3p Cys123Arg; Exon 8p Arg332X. These mutations have been described in the literature to be associated with the clinical phenotype of the proband and genetic investigation to the rest of the family was therefore recommended, but not yet performed.
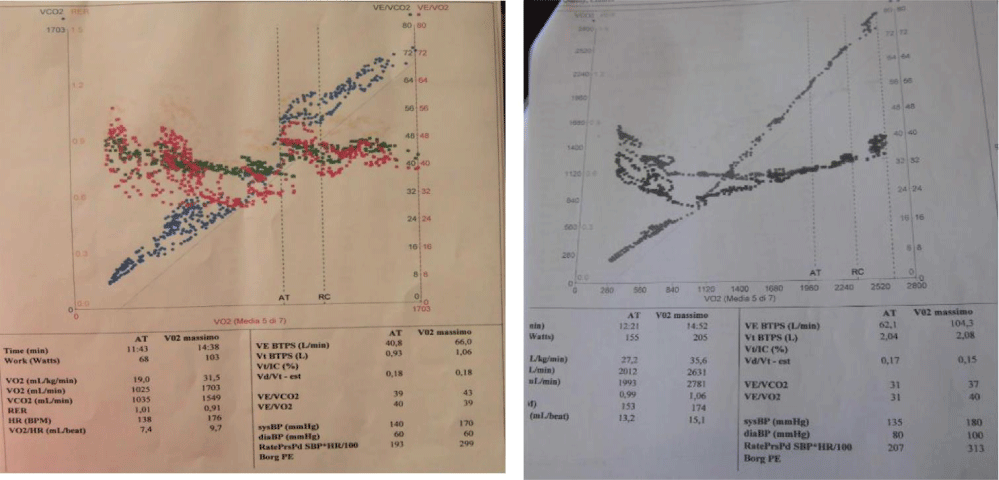 Figure 4: Case 1: CPET: VO2/HR 9.7 mL/beat; VE/VCO2 43; VE/VO2 39; VO2max 31.3 ml/kg/min; Case 2: CPET: VO2max of 35 ml/kg/min, VO2/HR 15 (ml/beat), VE/VCO2 37, VE/VO2 40.
View Figure 4
Figure 4: Case 1: CPET: VO2/HR 9.7 mL/beat; VE/VCO2 43; VE/VO2 39; VO2max 31.3 ml/kg/min; Case 2: CPET: VO2max of 35 ml/kg/min, VO2/HR 15 (ml/beat), VE/VCO2 37, VE/VO2 40.
View Figure 4
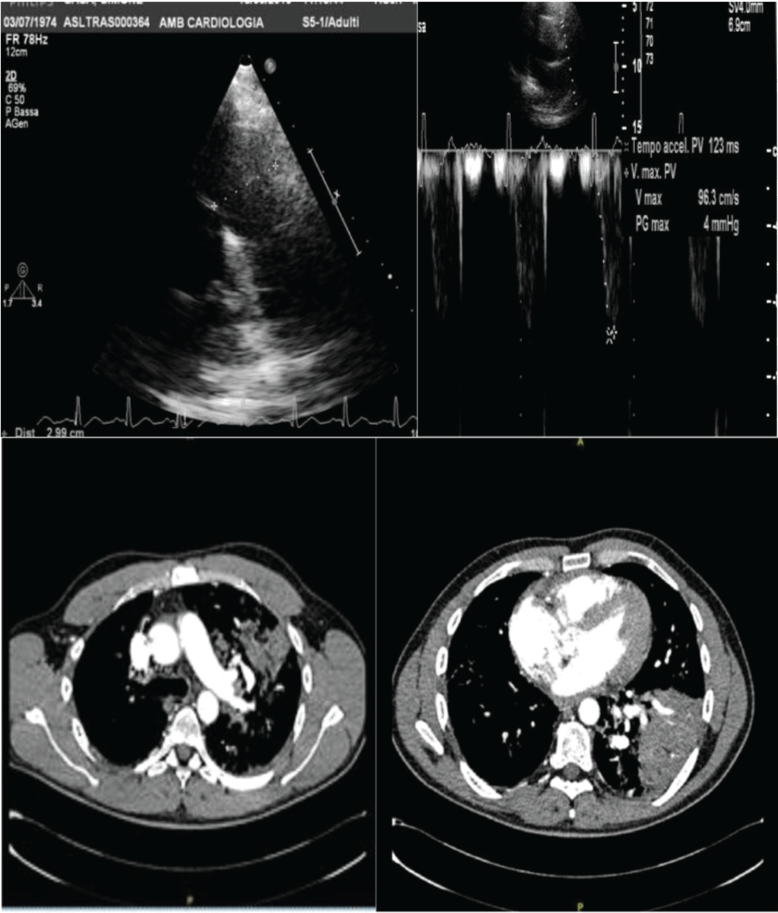 Figure 5: A) Case 2 echocardiogram: Pulmonary artery trunk and Actpo from parasternal views; B) CT Scan.
View Figure 5
Figure 5: A) Case 2 echocardiogram: Pulmonary artery trunk and Actpo from parasternal views; B) CT Scan.
View Figure 5
Unilateral PAA is a rare congenital condition that can be isolated or be associated more frequently with other conotruncal cardiac anomalies. This alteration comes from an embryogenetic defect in the sixth aortic arch of the affected side, causing the absence of the proximal segment of the ipsilateral pulmonary artery, which is associated with hypovascularization and hypoplasia of the lung of the same side [2,3]. In about 2/3 of the cases described the right pulmonary artery is affected and the agenesis of the left branch is frequently associated with other cardiovascular malformations. Mortality rate is about 7%, but both the presence of pulmonary hypertension and pulmonary bleeding phenomena can adversely affect the long-term prognosis [1,2]. In most cases, this condition is diagnosed in childhood, but some patients may remain mildly symptomatic or completely asymptomatic for many years and the diagnosis be quite incidental [4]. The most common clinical features include exertional dyspnoea, recurrent pulmonary infections or frequent asthmatic exacerbations, atypical chest pain, hemoptysis, pulmonary hypertension [2]. X-ray shows dislocation of the ipsilateral mediastinum and diaphragmatic elevation with volume loss of the affected lung, no hilar shadow and hyperinflation of the contralateral lung [5]. Chest CT scans confirm the suspected diagnosis by showing the absence of one pulmonary artery branch and the presence of atelectasic or bronchiectasic areas [6]. The echocardiogram and MRI scans allow excluding associated cardiovascular anomalies [2]. The angiography remains the method of choice for the diagnosis of pulmonary artery agenesis. But is reserved for cases where embolization for massive hemoptysis is indicated or for pre-operatory evaluation of patients undergoing surgical revascularization [4,7]. Treatment strategies of PAA depend very much on the clinical framework. Patients with hemoptysis, recurrent pulmonary infections are candidates for surgical treatment by pneumonectomy or lobectomy [4,7]. Embolization of bronchial systemic arteries is an alternative for patients with massive hemoptysis not eligible for surgery [4]. If pulmonary hypertension is present the options are revascularization of arterial missing branch or, if not feasible, treatment with specific medical therapy [1] as in the first case we reported.
PAA is a rare anomaly and the patient may remain asymptomatic till adulthood. Imaging plays a major role in the diagnosis and detecting the associated findings in heart and lungs. For patients who present with a unilateral small hemithorax, recurrent chest infections and an abnormal chest X-ray, to UAPA anomaly should be considered in the differential diagnosis. A careful diagnosis can avoid serious complications as pulmonary hypertension. Genetic screening should be performed.