The need for multiregional clinical trials for in Vitro diagnostic devices and extrapolating results in a new region is increasing in recent years. In this paper we are providing strategies for consideration of bridging studies for the two most common types of clinical trials for in Vitro diagnostic, method comparison and diagnostic clinical performance trials. Bridging studies should be considered when the expected agreement in the new region is inferior to the agreement in the original trial. In method comparison, a bridging study in the new region is not needed, if bias in this region is equal or smaller than the estimated bias of the original trial. Similarly, no bridging study is necessary for diagnostic clinical performance trials when sensitivity in the new region is equal to or greater than the sensitivity of the original trial. The reproducibility of the original trial has a significant effect on the sample size of the bridging study. Sample size decreases with the increase of reproducibility. We are using the ratio of reproducibility and the ratio of differences from the specification of the new region to the original trial to decide about the clinical trial in the new region.
Multiregional clinical trials have been used in the last decade or so to evaluate therapeutic products as part of developing global medicines. Approaches applied to these trials are summarized in the International Conference on Harmonization guideline ICH E5 [1], while several papers addressing bridging studies for therapeutic endpoints have laid the foundation for statistical procedures [2-4]. Recently, market requirements and regulatory landscape are changing for In Vitro Diagnostic (IVD) devices as well. Topics of multiregional clinical trials and bridging strategies were vastly discussed during the International Medical Device Regulatory Forum of China last year [5]. This forum organized by Chinese NMPA and aiming to address policies and regulations on medical devices, evidenced the increasing demand for multiregional clinical trials and bridging strategies to evaluate diagnostic products in new regions.
Accuracy performance is a way to evaluate IVD devices in clinical testing. ISO 5721-1 defines accuracy as the closeness of agreement between a test result and an accepted reference value [6]. The choice of reference depends on the diagnostic test and the type of device being tested. A newly discovered test is commonly evaluated for its accuracy to the clinical diagnosis. Clinical trials where diagnosis is used as a reference method are called 'diagnostic clinical performance' trials and accuracy is commonly evaluated in terms of sensitivity and specificity [7,8]. For other IVD tests, a marketed device can serve as a predicate for the candidate device under test and agreement with the comparative method is evaluated based on the closeness of the agreement of the measurements. These types of clinical trials are referred to as 'method comparison'. Bias between devices is an evaluator of agreement that can be estimated statistically by using a regression approach [9].
Shao & Chow [10] and Chow, et al. [3] introduced the concept of reproducibility to assess the ability of the results from a clinical trial to be extrapolated to a new region. Quan, et al. [11] and Ikeda & Bretz [12] provided approaches for sample size calculation for bridging study in a new region using the concept of consistency defined earlier by Shih [2]. Lan, et al. [13] used weighted z-statistics to design and size bridging studies while Liu, et al. [14] proposed a Bayesian approach using the prior information to assess the similarity between the original clinical trial and bridging studies in the new region. All these methodologies were developed for therapeutic endpoints, i.e., comparing test results to a placebo, survival endpoints, etc. We are not aware of any publication addressing bridging methodologies to support the introduction of IVD products into new geographies or region.
Consequently, the purpose of this paper is to provide a strategy for bridging the results of an original clinical trial into a new region for IVD devices. We will consider the two most common types of clinical trials for IVD, method comparison and diagnostic clinical performance trials.
This section is only for method comparison studies for quantitative measurement procedures. Let X be the measurement with the predicate device and Y the measurement with the test device. Both devices are tested in N subjects, with Xi and Yi being the measurement of both predicate and test devices, respectively, on the same i subject.
The linear function (1) fits the relationship between devices.
Y = b0 + b1X + ε (1)
Where b0 and b1 are intercept and slope, and ε is the error.
Since both test and predicate devices measure with a certain degree of imprecision, Linnet [15] proposed an orthogonal least square approach to estimate parameters of (1). This approach is usually referred to in the IVD literature as Deming approach [9]. Non-parametrical estimation of the parameters is also commonly used in the IVD industry [9,16].
Bias (B) between two methods at a certain medically important level Xc can be calculated as:
B = b0 + (b1 - 1) Xc (2)
The standard error of B can be calculated based on ordinary least square variances and covariance of the slope and intercept. Linnet [17] argued that for a relatively large analytical measuring interval (AMI), slope close to 1 and equal precision of both devices, orthogonal least square estimate of var(B) can be approximated by its ordinary least square estimate. Considering variances-covariance of the regression estimates, se(B) can be approximated as:
Where σ is the precision and is the average of the predicate device. se(B) is proportional to precision, AMI, as well as the distance of Xc from the mean. The function under the square root is multiplied by 2 since both devices are measured with error. Knowledge about the precision of the device, AMI of the analyte, and a prespecified clinical tolerable bias (Bs) can be used to prospectively calculate power and size the clinical trial.
Hypotheses for equivalence between the predicate and test device in method comparison are:
H0: B ≤ -Bs or B ≥ Bs
H1: -Bs < B < Bs
Prespecified bias, Bs is usually symmetrical around zero. The absolute value of the bias represents the clinical relevance for an analyte while the sign indicates the direction of the bias. However, in specific comparisons, bias might be asymmetrical.
Let P be the prospective power to size the original clinical trial. Let and be the estimated bias and standard error from the original clinical trial, and tc a statistic that follows a t-distribution with (N-2) degree of freedom.
Let be the post experimental power of the original trial. We can use Owen's function as [18]:
Where t is from t-distribution with 1-α confidence and n-2 degrees of freedom, Φ(.) is the cumulative distribution function and (.) is the density function of the standard normal distribution, while,
Power can be calculated as the difference:
Shao and Chow [10] defined post experimental power as reproducibility probability of the original clinical trial .
When devices are tested on a different set of subjects in the new region, the estimate of bias might be different from the original trial due to population differences. Let denote the estimate of bias in the new region and denotes the estimate of standard error. The values of these estimates can be based on knowledge, if available, or several different values can be used in the calculation to perform a sensitivity analysis. Difference between the estimates in the original clinical trial and the new region clinical trial is defined by Chow, et al. [3] as the similarity index (Δ). In case of method comparison, = since precision and AMI are characteristic of the device they do not depend on the population. Consequently, Δ is only a function of the difference in bias.
Let and Equation (4) can be used to calculate the reproducibility probability of the trial in the new region, Results of the original region can be extrapolated to the new region when When a bridging study in the new region is not required [3].
Let and Consistency between the results of the original trial and the expected results in the new region can be expressed as [12]:
A consistency probability of γ ≥ 0.8 and the values of ρ ≥ 0.5 are generally acceptable. When D and Dnew are normally distributed, Ikeda & Bretz [12] showed that equation (5) can be approximated by a normal cumulative density function and the proportion of subjects in the new region can be calculated as:
where and α = 0.025 for 95% confidence. Since the sensitivity of using different values of is evaluated, both and p represent vectors of values. The number of subjects in the new region is:
Example
A clinical trial to comply with the pre-market regulatory requirement for testing Beckman Coulter DxH 520 hematology analyzer was conducted at different sites in the United States. DxH 520 was the test device while UniCel DxH 800 Coulter Cellular Analysis System was the predicate device. Hematology analyzers measure several hematology parameters at the same time. In the context of this example, we will focus on only one parameter, platelet (PLT) which is a cell type related to blood coagulation. PLT expressed as cell counts/μL × 103 was measured by both devices on N = 196 subjects. Because of a large number of counts for each measurement, both Xi and Yi are assumed to be normally distributed, with means μx and μy and the same precision, σ.
Method comparison was used to evaluate the agreement and establish equivalence between devices. PLT counts in humans exhibit relatively wide range depending on clinical conditions. This can affect the variability of measurements throughout the AMI. In addition, the relative variability (CV%) of precision in the low range is much larger in comparison to the rest of AMI. Consequently, there is a pattern of dependency of variability within the AMI that required weighted adjustments for the estimation of regression parameters.
Weighted Deming approach [15] was used to estimate regression parameters. The estimated slope was 1.02 (95% CI; 0.97, 1.06) and intercept was -4.86 (95% CI; -15.21, 5.49). There are two medically important levels for PLT; Xc = 150 cell/μL × 103 and Xc = 450 cell/μL × 103. Biases at these levels were calculated using equation (2) while a jackknife resampling was used to estimate The two-sided confidence intervals of were calculated as (α = 0.05 and N = 196):
A 10% bias is usually tolerable for PLT. Consequently, Bs = 15 cells/μL × 103 at Xc = 150 cells/μL × 103 and Bs = 45 cells/μL × 103 at Xc = 450 cells/μL × 103. Results are provided in Table 1.
Table 1: Bias at two different medically important levels. View Table 1
Using this information and equation (4), The value of Δ in the new region is unknown. We calculated for different values of Δ in the interval The respective values of were also calculated for the same interval. The summary of the results for Xc = 150 are shown in Table 2. No clinical trial is necessary in the new region when bridging study is recommended when and ρ ≥ 0.5, while a fully powered new clinical trial is needed when ρ < 0.5. A different set of Δ can be used if there are no solutions for one of the above conditions. Based on PLT results, a bridging study is recommended only when Δ = 6 cells/μL × 103 that correspond to a sample size of 18 (Table 2).
Table 2: Decision for a method comparison trial in the new region (Xc = 150 cells/μL × 103). View Table 2
Decision for a bridging study corresponds to Δ = 6 for Xc = 150 (Table 2) and the fraction of the difference from the prespecified clinical tolerable bias is This fraction corresponds to Δ = 18 for Xc = 450. Calculation for and ρ and decision are shown in Table 3 starting from Δ = 17. All decision results for a reasonable interval indicate that no clinical trial is need in the new region.
Table 3: Decision for a method comparison trial in the new region (Xc = 450 cells/μL × 103). View Table 3
Based on the results from the two medically important levels, is it evident that bias at Xc = 150 level represents the worst-case scenario and drives the decision on the clinical trial. The relative value of this bias is 1.24% compare to 0.92% at Xc = 450.
Let C be a binary variable denoting the true clinical status for a disease. C = 1 when the disease is present and C = 0 when the disease is absent. Let X be the results of an IVD test, where X = 1 indicates the presence of the disease while X = 0 indicates the absence of the disease. X can have a binary outcome or can be measured on a continuous scale. When measured on a continuous scale, X can be transformed into binary by using a cut-off point that separates the values of X in two categories, disease present or absent.
Comparison of a test result to clinical truth can have four outcomes; True positive = (C = 1, X = 1), False negative = (C = 1, X = 0), False positive = (C = 0, X = 1) and True negative = (C = 0, X = 0). Let N be the total number of subjects in the trial.
N = NTP + NFN + NFP + NTN
where NTP, NFN, NFP, and NTN are the numbers of true positive, false negative, false positive and true negative respectively. Based on these outcomes, several conditional probabilities can be calculated to evaluate the diagnostic ability of the test [19]. The most common are sensitivity, specificity, positive/negative predicted values, positive/negative likelihood ratios, etc. In this section, we will focus on sensitivity, which is the proportion of subjects with disease that are tested positive. The same approach can be used for specificity and predicted values.
S = p (X = 1 | C = 1)
Let S be a target value for sensitivity and Ss a lower limit of sensitivity that is clinically acceptable. Prospective power (P) can be directly calculated from the cumulative normal distribution function as:
While the required number of diseased subjects for the original clinical trial is:
Let be the estimated sensitivity from the original clinical trial.
is the standard error of a binomial proportion, while normal approximation can be used to calculate confidence intervals. Post experimental power is:
The similarity index is only a function of differences in sensitivity between the original clinical trial and the expected estimates in the new region, Δ = S - SNew, while can be calculated based on the power function (9), S and Δ. When D = S - Ss and Dnew = SNew - Ss, consistency between the results of the original trial and new region and proportion of subjects in the new region can be calculated based on equations (5) and (6).
Example
Data from the sepsis clinical trial presented in Crouser, et al. [20] were used for demonstration purposes. Clinical trial conducted at three different sites in the US, enrolled NDisease = 385 sepsis positive subjects out of a total of N = 2158 subjects. Monocyte distribution width (MDW) was measured and evaluated for the ability to diagnose sepsis. The MDW was measured on a continuous scale and the use of a cut-off (Xc = 20) separated the values into X = 1 when MDW > 20 and X = 0 when MDW ≤ 20 [20].
Sensitivity; (95% CI; 0.69, 0.78) based on NTP = 285 and NFN = 100. The lowest clinically acceptable value for sensitivity was Ss = 0.65. Based on this information reproducibility of the original trial was Since the value of Δ in the new region is unknown we choose a series of values in the interval Ss ≤ Δ ≤ and ρ are also calculated for the same interval of Δ values. The results are shown in Table 4. At least 78 sepsis positive samples are needed in the new region.
Table 4: Decision for a diagnostic clinical performance trial in the new region (sensitivity). View Table 4
Specificity; (95% CI; 0.69, 0.74) based on NTN = 1275 and NFP = 498. The lowest clinically acceptable value was Ss = 0.65. Results are shown in Table 5. At least 122 negative samples are needed in the new region.
Table 5: Decision for a diagnostic clinical performance trial in the new region (specificity). View Table 5
The performance in the original trial expressed in terms of reproducibility has a significant effect on sample size of bridging study. To demonstrate this, we simulated data calculating sample size for bridging study by varying and ρ. The proportion of the sample size was calculated for the significance level of α = 0.05 and consistency of γ = 0.80. Results in Figure 1 and Table 6 show the proportion of subjects (ρ) required for the different threshold of when in the original region varies. Each curve in Figure 1 corresponds to a specific reproducibility value.Values of ≥ 0.8 are selected for these simulations because multicenter trials for IVD are usually prospectively powered at P ≥ 0.8 level. The proportion of the subjects increases with the increase of ρ, however, decreases with the increase of the reproducibility of the original trial.
Table 6: Simulated proportion of subjects. View Table 6
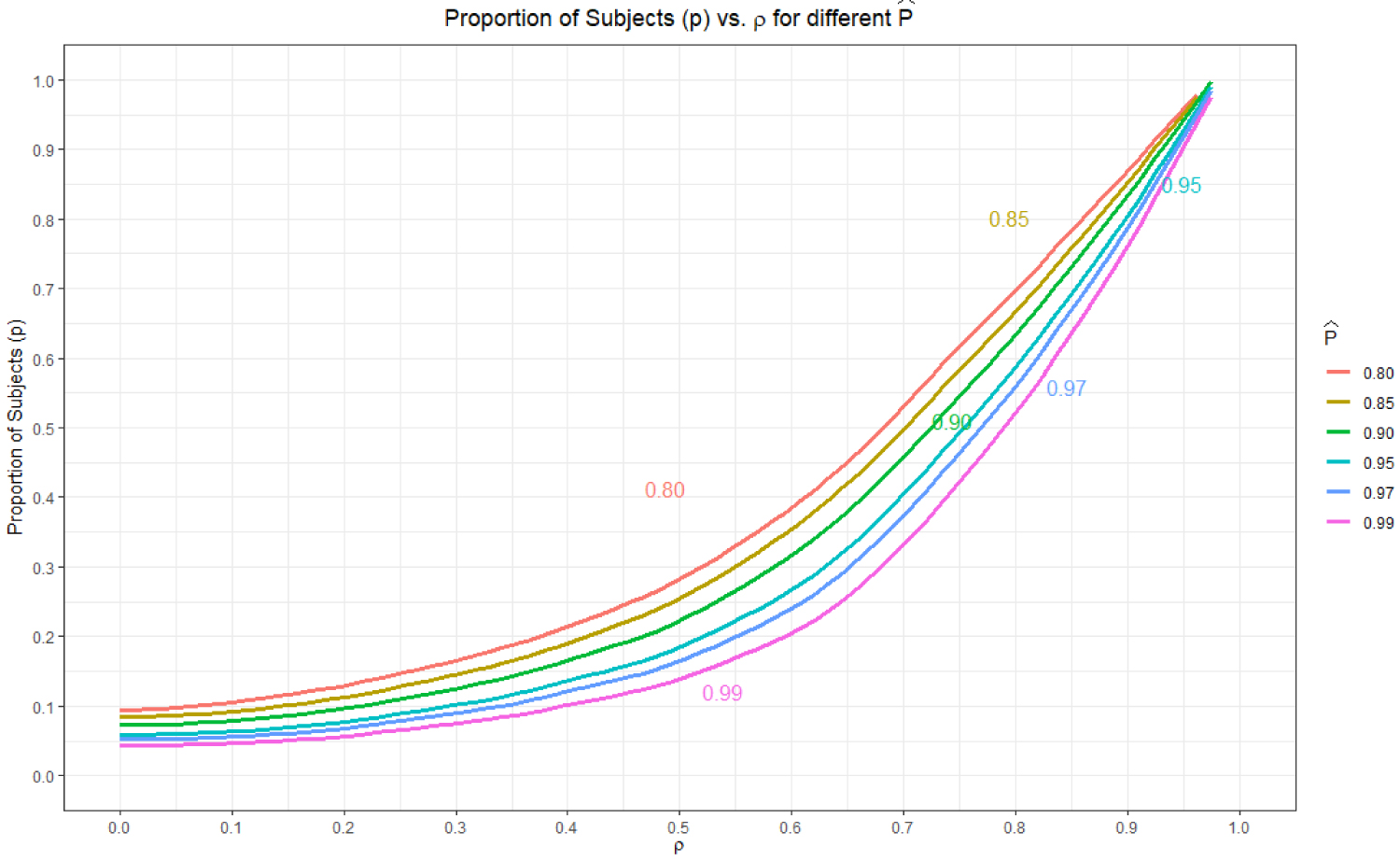 Figure 1: Proportion of subjects in the bridging study.
View Figure 1
Figure 1: Proportion of subjects in the bridging study.
View Figure 1
The relationship between and ρ is shown in Figure 2. Values of ρ < 0.5 are not possible when substantiating the decision of no need for bridging study when only The value of ρ that corresponds to is approximately 0.75 and bridging is necessary when 0.5 ≤ ρ ≤ 0.75. From Figure 1, we also see the most separation of the sample sizes for different occur in the 0.5 ≤ ρ ≤ 0.75 interval. Consequently, any bridging study decision and subsequent sample size calculations should be in the region and ρ ≥ 0.5.
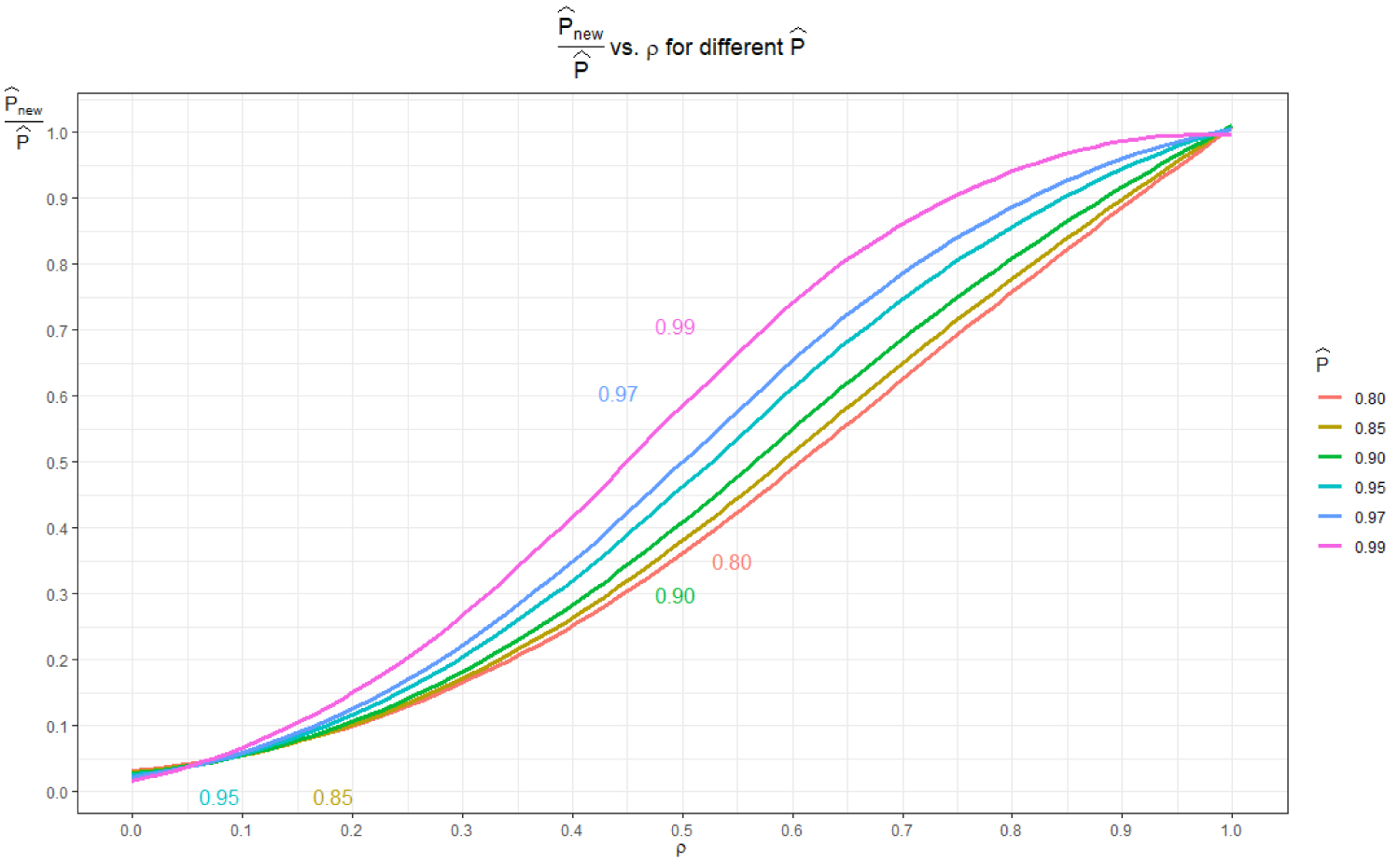 Figure 2: Relationship between and ρ.
View Figure 2
Figure 2: Relationship between and ρ.
View Figure 2
There are three available options when determining whether to conduct a clinical trial in the new region; extrapolate the results of the original trial to the new region (no need for a new clinical study), perform a bridging study, and conduct a new fully powered clinical trial. The decision about these choices is contingent on the difference in agreement between the original trial and the expected agreement in the new region. We also showed that the reproducibility of the original trial significantly affects sample size of the bridging study.
The reproducibility of the original trial is related to the prospectively calculated sample size and estimated agreement performance in the trial. Sample size should be based on at least 80% power (90% is preferred for method comparisons) while estimated performance depends on the product being tested. Prospectively powering a method comparison trial separately for slope and intercept has been discussed by Linnet [17] and Passing & Bablok [21]. We are showing an approach to approximate standard error of bias based on variance-covariance structure of slope and intercept and use the knowledge about bias acceptance limits, precision, and measuring interval to calculate sample size for a predetermined power and confidence level. Other approaches for evaluating agreement in method comparison clinical trials are not discussed in this paper [22,23]. Powering of diagnostic clinical performance trials is based on normal approximation of a binomial proportion. Approaches like the exact binomial calculations [24], powering both sensitivity and specificity Pepe [19], and others are not considered here but might be subject to future publications.
Bridging studies should be considered when the expected agreement in the new region is inferior to the agreement in the original trial. In method comparison, a bridging study in the new region is not needed, if bias in this region is equal or smaller than the estimated bias of the original trial. Similarly, no bridging study is necessary for diagnostic clinical performance trials when sensitivity in the new region is equal to or greater than the sensitivity of the original trial.
We are using the ratio of reproducibility and the ratio of differences from the specification (ρ) of the new region to the original trial to decide about the clinical trial in the new region. Shao and Chow [10] recommended a 90% level of reproducibility ratio as a cutoff level for considering a bridging study in a new region. Values of ρ ≥ 0.5 are based on the Japanese Ministry of Health, Labor, and Welfare guidance and are used in several publications [11,12,25]. Both these values are related to therapeutic drug testing and authors recommend modifying them according to the specific product being tested.
There is no historical evidence or literature that we know about using these values to design bridging studies for IVD. We are recommending the following steps:
1. Estimate reproducibility of the original trial
2. Simulate a range of expected differences in the new region (Δ)
3. Estimate reproducibility for each of these differences in the new region
4. Calculate the ration of reproducibility in the new region versus the original trial ()
5. Calculate
6. Guide to inform decision:
a. No need for bridging study when
b. Consider bridging study when and ρ ≥ 0.5
c. New clinical trial when ρ < 0.5
Sometimes there might be no solutions for the combination of values above. In these cases, a new set of expected differences in step 2 will need to be simulated. We also caution that the values of ρ > 0.6 could require the bridging study to be too large and not practical. Ko, et al. [25] also argued that larger values of ρ would indicate that the overall results are dominated by the new region and consistency of the results between the original trial and the new region might not be valid. For these reasons, we recommend that the sample size for bridging study be based on the minimum proportion in the interval of, and ρ ≥ 0.5.
Bias in method comparison studies might be calculated at different medically important levels. We recommend identifying the highest relative bias to the prespecified clinical tolerable bias at a medical level and make decision for bridging studies for that bias. In addition, some IVD devices provide measurements for multiple analytes. In these cases, we recommend that calculations be performed for each analyte separately and decision be made based on the worst performing analyte. Similarly, both sensitivity and specificity need to be considered in diagnostic clinical performance trials.
Sample size for bridging study represents the minimum sample size to satisfy the pre-determined requirements and conditions. However, if necessary, more subjects can be tested to uniformly cover the AMI.
Like other authors in the therapeutic domain, we recommend that the choice of and ρ to be based on the clinical relevance of IVD under test as well as regulatory requirements in the new region. In addition, recommendations for bridging studies in this paper are based on statistical considerations. Other factors may influence the final decision.