Acute kidney injury (AKI) represents a great challenge for the anesthesiologist during the perioperative period, since its presence directly impacts in patients mortality and morbidity, even after its resolution as a result of multiples factors. Furthermore, it is characterized by coexisting with a great variety of systemic alterations, which add special difficulty to its study and understanding. During perioperative period there may be ischemic insult and non-ischemic insult, on the other hand, volume overload (hypervolemia) can also cause alteration on kidney function by modifying the consumption and delivery of renal oxygen. Throughout, some of the most important points for understanding this topic are presented in the present paper.
Acute kidney injury, Perioperative, Hypervolemia
Acute kidney injury (AKI) is the most frequent syndrome in hospitalized patients, its prevalence has been measured from 15% to more than 50% depending on the statistics of each service, whether chest surgery, oncology, transplants or intensive care units [1]. It is characterized by coexisting with a great variety of systemic alterations, which add special difficulty to its study and understanding, making individualized treatments necessary in each of its presentations: Cardiorenal syndrome [2], hepatorenal syndrome [3], nephrotoxicity [4] or sepsis associated [5]. However, the most important thing, indubitably, is that its presence impacts the prognosis of the patient through an increase in morbidity and mortality which persists even after the resolution of the AKI itself [6], this can be explained by factors such as: Endothelial dysfunction [7], myocardial remodeling [8], increased oxidative stress [9] and epigenetic factors [10]. The purpose of this study is to describe renal oxygen metabolism, as well as to explore the pathophysiology of kidney damage and its possible relationship with fluid therapy in a perioperative setting.
The diagnosis of AKI has never been easy, in fact, there is currently a lack of a "gold standard" for this purpose, however, in 2004 the ADQI (Acute Dialysis Quality Initiative) proposed for the first time a consensus diagnostic criteria called RIFLE (Risk, Injury, Failure, Loss, End-stage renal disease) for the detection of AKI, based on creatinine levels, urinary volumes and evolution time [11]; later in 2007 these criteria were modified by the Acute Kidney Injury Network (AKIN), whose main contribution was the discovery that small changes in plasma levels of creatinine (Cr) can affect the prognosis of patients [12] and, finally, the Kidney Disease Improving Global Outcomes (KDIGO) introduced in 2012 gave the criteria that are currently in use [13], including relative and absolute changes in plasma Cr levels with respect to time; either short term (48 hrs) or long term (> 7 days). Table 1 makes a comparison of the three classifications. In recent years, numerous biomarkers have been proposed in addition to creatinine for the diagnosis of AKI, with the purpose of establishing a timely treatment that improves the prognosis. Among the mentioned biomarkers we can find: Neutrophil gelatinase-associated lipocalin (NGAL), cystatin C, kidney injury marker 1 (KIM-1), N-acetyl-beta-D-glucosaminidase (NAG), tissue inhibitor of metalloproteinases 2 (TIMP-2) and Insulin-like growth factor-binding protein (IGFBP-7). These molecules all have different features, and we provide a summary of them in Table 2 [14-18].
The development of AKI in the perioperative setting can be the consequence of multivariate causes, however, the most evident is the transitory interruption of renal blood flow (RBF), which has been identified mainly in major cardiac surgery [19]. The kidneys receive 20-25% of the total cardiac output (CO) of the human body, approximately between 1,000 and 1,200 milliliters of blood per minute, oxygen is distributed in a heterogeneous manner, mainly through the cortex where it reaches a concentration 10 times higher than in the renal medulla. Faced with an abrupt transitory drop in RBF, intrinsic and neurohumoral self-regulatory mechanisms cause proportional changes in glomerular filtration in a way that can lead to the diagnosis of AKI [20]. This can occur as a consequence of 1) Hypovolemia, 2) Hypotension, 3) Renal venous congestion [21-23].
Whenever this drop in RBF is short enough, it does not cause significant alterations in the homeostasis of the tubular segments of the nephron, which is why it is considered a prerenal or hemodynamic AKI. However, when this hypoperfusion lasts long enough, it will cause ischemic cellular damage, mainly in the tubular segments with higher metabolic requirements and lower oxygen supply: The straight portion of the proximal tubule and the thick ascending portion of the loop of Henle, both located in the external medulla [24]. Different experimental models have made it possible to understand the biological mechanisms that develop during AKI, with the mouse renal ischemia-reperfusion injury models (I/R) acquiring greater relevance in order to understand the perioperative scenario, establishing a clearer temporality in comparison with other AKI syndromes [25,26]. Thus, the processes of mitochondrial injury, endoplasmic reticulum stress and cell death that occur as a consequence not only of oxygen deficiency, but also due to oxidative stress during reperfusion have been recognized [24,27,28].
During the first 72 hours of reperfusion, the injured kidney presents gradual morphological and functional changes that are characterized by a loss of polarity of the tubular cells, tissular death due to apoptosis and necrosis, and the establishment of a significant inflammatory infiltrate [29]. One of the most recognized adaptive responses during this phase is mediated by the hypoxia-inducible factor (HIF), which is a stabilized heterodimer during hypoxia, due to the fact that under normoxic conditions, the alpha subunit is hydroxylated by different oxygenases, which induces its proteasomal degradation through the Von Hippel Lindau protein [30]. An increase in anaerobic metabolism, angiogenic gene expression, and inflammatory regulation are some examples of the pathways activated by the HIF [31,32]. Several experimental studies have proven a crucial role of HIF-alpha in the kidney response to hypoxia, particularly as related to a secondary to damage due to I/R, facilitating survival of the tubular epithelium, regulation of interstitial inflammatory response and maintaining endothelial integrity [33-36].
Another important mechanism in the renal response to ischemic insult is the activation of the renin-angiotensin-aldosterone system, given that after a hypoperfusion stimulation, the renin secretion and intrarenal generation of angiotensin II favor oxidative stress in tubular cells, endothelial dysfunction and proinflammatory signaling both in the parenchyma and in infiltrating cells [37]. Furthermore, aldosterone-dependent and independent mineralocorticoid receptor activity also contributes to establishing kidney damage [38,39]. In fact, it has been shown that prophylactic administration of losartan, spironolactone or even bilateral adrenalectomy 3 days before the ischemic insult is capable of significantly reducing cellular, vascular and inflammatory damage in rats [40-42]. Finally, the activity of immune cells attracted by damage-associated molecular patterns (DAMP) and different cytokines secreted in the renal parenchyma also play an important role in establishing acute kidney damage through the activation of pro-apoptotic inflammatory pathways where neutrophils, macrophages, T lymphocytes, endothelial cells, and the renal tubule itself take a part in [43].
There are other factors other than I/R that can determine the establishment of AKI in a surgical patient, among which we can note the exposure to nephrotoxic drugs, and septic syndromes [44]. Nephrotoxic damage depends on two factors: 1) The properties of the drug and 2) The biological predisposition of the patient. There are many commonly used drugs with nephrotoxic effects (as listed in Table 3). In a general manner, the following points are recognized as kidney injuries caused by nephrotoxic drugs effects [45]:
a. Prolonged renal exposure to the drug.
b. Active drug transport in the renal epithelium, competing with other solutes.
c. Cytosolic accumulation in inclusion bodies or in membranous organelles.
d. Generation of crystals or casts due to highly insoluble urine or interaction with proteins such as uromodulin.
e. Induction of immunological activity.
On the other hand, the presence of infectious processes, mainly sepsis, can cause kidney damage through mechanisms independent of the surgical insult, mediated by renal vascular dysfunction, tubular response to pathogenic toxins, with important metabolic alterations and through an increase in inflammatory activity within the parenchyma. These factors can worsen the prognosis of the patient undergoing surgery, and whenever added to a risk of developing AKI due to I/R, they may contribute significantly to the establishment of kidney damage and an increase in the probability of negative outcomes, such as mortality or irreversible loss of kidney function [45].
Fluid therapy is essential in the management of patients during the perioperative period; in fact, it is the second most used therapy in critically ill patients [46] with the aim of improving hemodynamics and tissue perfusion [47], despite the fact that the correlation between hypovolemia and hypoperfusion seems clear in the development of AKI, there is evidence in animal models that suggests that the increase in RBF might also lead to kidney damage [48,49], therefore, fluid therapy is not without its complications and that is why intravenous solutions should be construed as what they really are, drugs with unique pharmacokinetic and pharmacodynamic features, adverse effects and even lethal doses [50,51], a view that has been named the four Ds of fluid therapy: Drug, Dosing, Duration and De-escalation [52]. It is a common practice in anesthesiology to promote a certain degree of hypervolemia after anesthetic induction with the purpose of maintaining organic perfusion during the surgical procedure [53], a rationale known as "wet" which is considered the gold standard of perioperative fluid therapy [54]; however, providing fluids in this period of time will only cause a state of hypervolemia, since a large part of the administered fluid will remain in the venous system as a consequence of its large capacitance, making it necessary to administer more fluid in order to achieve a transitory blood pressure increase and, thus, creating a vicious circle once the administered fluid has been redistributed with the consequent decrease in blood pressure [55,56], since the cause of hypotension is not intravascular volume, but a vasodilation that is secondary to the anesthetic drugs. Additionally, one must take into account that there are three variables that might assist us in explaining the effect of fluid therapy on organ perfusion; the functional capillary density (FCD), which refers to the capacity of the microcirculatory diffusion of oxygen; the erythrocyte velocity (RBCv), a measurement of the convective capacity of the microcirculation capillaries to diffuse oxygen and the capillary hematocrit (cHct), which is an hematocrit value in the capillary as it relates to time [57,58].
The administration of fluid therapy will affect DF, RBCv and cHct causing a loss of hemodynamic coherence mainly by means of two mechanisms [59]. The first, hemodilution, causing a decrease capillary erythrocytes concentration and an increase in the diffusion distance between erythrocytes and tissue, affecting the oxygen transport capacity [60], another consequence will be alterations in the viscosity, noted by a decrease of the same accompanied with loss of shear stress, which is essential for vasoregulation [61]. The second mechanism is the formation of reflex edema due to a capillary leak related to endothelial damage (glycocalyx smooth) with the subsequent increase in the diffusion distance, which in turn is the result of interstitial water accumulation and hemodilution [59]. The damage to the glycocalyx is explained by the fact that, whenever there is a state of hypervolemia, the same will be stopped by the cardiac fiber, causing the release of natriuretic peptides (NP) as a compensatory mechanism in order to stimulate natriuresis [62]. Both the NPs and the administered fluid will cause damage to the glycocalyx [63], first through a degradation mechanism, suggested by an observed increase of molecules such as syndecane or hyaluronan [64,65], the second, by a direct damage mechanism acting on the vascular endothelium, caused by the increased shear stress [63]. In addition to the aforementioned, there is a paradoxical effect in which the increase in blood volume will in turn increase the metabolic consumption of renal oxygen; given that increasing the RBF will, in turn increase the GFR with the purpose of maintaining body's homeostasis, which will be translated, paradoxically, as an increase in renal oxygen consumption (VO2r), all of which will be offset by an increase in renal oxygen extraction (ExO2r), explaining, therefore, the limited renal capacity to raise its DO2r as it increases RBF (Figure 1) [66].
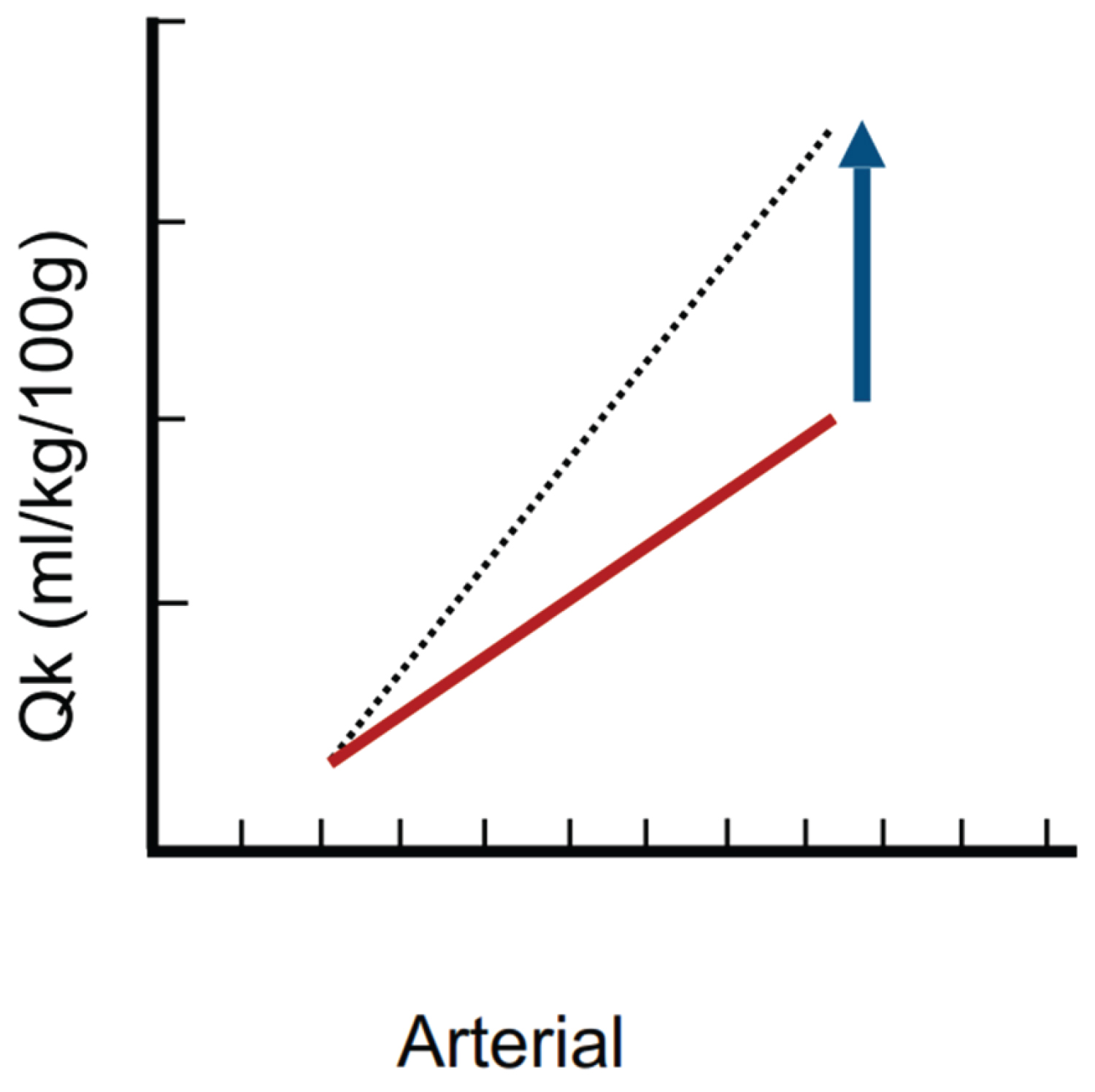 Figure 1: Kidney flow-pressure graph. The solid line indicates the baseline state, meanwhile the renal has a steep slope explained by the high amount of cardiac output it receives and the difference between both lines represents the functional reserve that determines the renal capacity to couple the blood flow for a given arterial pressure, which is very limited. Modified image Magder S [55].
View Figure 1
Figure 1: Kidney flow-pressure graph. The solid line indicates the baseline state, meanwhile the renal has a steep slope explained by the high amount of cardiac output it receives and the difference between both lines represents the functional reserve that determines the renal capacity to couple the blood flow for a given arterial pressure, which is very limited. Modified image Magder S [55].
View Figure 1
AKI is one of the most frequent complications in many clinical settings, playing an important role in the impact of morbidity and mortality of patients. Perioperative scenarios are no exception; therein, as in other scenarios, lies a pool of probable multifactorial origins: Hypovolemia, hypotension, non-ischemic insults or even hypervolemic states (venous congestion), which may be the culprits. Currently, anesthesiologists must be able to recognize all these possible causes and, since there is currently no precise maneuver that ensures complete abolition of these complications, a global approach will be the best option for managing patients that undergo anesthetic-surgical procedures.
The authors declare no competing interests.