Adult-Onset Still’s Disease (AOSD) with Secondary Hemophagocytic Lymphohistiocytosis (HLH) is an autoimmune disorder that is most commonly a diagnosis of exclusion. AOSD is a known trigger of secondary HLH, amongst other inflammatory and infectious processes, making the association between the two already rare diseases even more intriguing. It commonly presents with non-specific symptoms like fevers, myalgias, night sweats, chills, paresthesias, and is associated with a relapsing-remitting salmon colored rash. Our patient is a young African American woman in her 20s who was diagnosed with AOSD with secondary HLH after meeting the Yamaguchi Criteria for AOSD and the HLH-2004 criteria for HLH, both being the gold standard criteria for the respective diseases.
Adult-Onset Still’s Disease (AOSD) is a rare inflammatory disorder typically characterized by relapsing and remitting fever, polyarthritis, and transient cutaneous maculopapular rash. AOSD presentation is rare and often a diagnosis of exclusion. An identifying feature of this condition revolves around the markedly elevated ferritin levels often greater than 1000 ng/mL. AOSD is typically a top differential after patients present with the typical symptoms in “Still’s triad,” arthritis, evanescent salmon-colored rash, and quotidian fevers [1]. Although AOSD is already a rare diagnosis, further variations can occur. Hemophagocytic Lymphohistiocytosis (HLH) is an autoimmune inflammatory disease that causes severe inflammation and tissue destruction secondary to the absence of normal down regulation of activated macrophages, natural killer cells, and cytotoxic lymphocytes [1]. Similarly, Macrophage Activation Syndrome (MAS) is an overlapping diagnosis that is equally destructive to HLH. However, both of these severe processes are triggered by a primary acute inflammatory state. MAS is classically associated with rheumatologic conditions such as systemic juvenile idiopathic arthritis (sJIA) and adult-onset Still’s disease (AOSD). Whereas HLH is associated with viral infections, malignancy, and certain chemotherapies [2].
This case, specifically, describes AOSD with Secondary HLH as it relates to a previously healthy patient. Moreover, this case describes the process of diagnosing AOSD with Secondary HLH in a young female patient who presented with nonspecific flu-like symptoms including myalgias, intermittent fevers, night sweats, and a relapsing-remitting rash.
An African American woman in her 20s with a past medical history of anxiety initially presented with nuchal rigidity and myalgias. On admission, the patient reported body aches, intermittent fevers (Tmax = 104 ° F), night sweats, chills and “pins and needles” sensation in bilateral upper extremities. Moreover, the patient experienced a relapsing and remitting urticarial rash without a known trigger or anaphylaxis.
Prior to her hospital presentation, the patient had an unremarkable outpatient workup to identify a specific allergic etiology but was prescribed a short steroid dose pack and steroid cream with significant relief of her symptoms and rash. Two weeks prior to this admission, she was noted to have a separate admission for myalgias and intermittent fevers, for which she was treated with intravenous fluids. At that time, her infectious workup was negative including negative blood cultures. She was ultimately discharged with PCP follow up. Additionally, she had recent travel to Miami and Kansas City but denied any other symptoms including headaches, syncope, nausea, vomiting, or shortness of breath.
Once admitted, she was started on empiric ceftriaxone, vancomycin and acyclovir for suspected meningitis and appropriate workup was initiated. Patient was persistently alert and oriented to person, place, time, and situation. On admission, her physical exam was significant for reduced cervical flexion due to pain, minimally palpable posterior cervical lymphadenopathy. However, the physical exam was not remarkable for Kernig or Brudzinski signs.
Table 1 details relevant admission labs below including ferritin levels were elevated at 9358 ng/mL. Lactate dehydrogenase elevated at 946 U/L. Decreased TIBC and Transferrin at 170 ug/dL and 134 mg/dL, respectively. CT imaging of the head and cervical spine was unremarkable for any acute intracranial process or acute osseous cervical spine abnormalities.
Table 1: Pertinent admission labs. View Table 1
Over the course of the patient’s admission, she reported transient subjective fevers and chills with associated night sweats every evening between 6:00 PM - 7:00 PM. Her rash reappeared twice, once on her anterior right thigh and once on her posterior left thigh (see Figure1, Figure 2, Figure 3 and Figure 4).
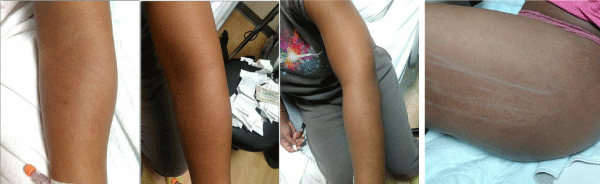 Figure 1-4: Salmon colored, relapsing-and-remitting urticarial rash over left arm and leg.
View Figure 1-4
Figure 1-4: Salmon colored, relapsing-and-remitting urticarial rash over left arm and leg.
View Figure 1-4
On day 3 of hospitalization, her nuchal rigidity resolved, however, she acutely developed shortness of breath and pleuritic chest pain relieved by position, which was concerning for pulmonary embolism and acute pericarditis. At this point, the patient satisfied SIRS Sepsis criteria overnight with a temperature of 100.8 F, HR 125 and MAP of 57 mmHg, and treatment was initiated. She also met acute pericarditis diagnostic criteria when she developed positional nonischemic chest pain, and an echo showed a small pericardial effusion without tamponade (see Figure 5) and an EF of 65%. Her EKG revealed new T wave inversions in V2-3 and the troponin panel was negative.
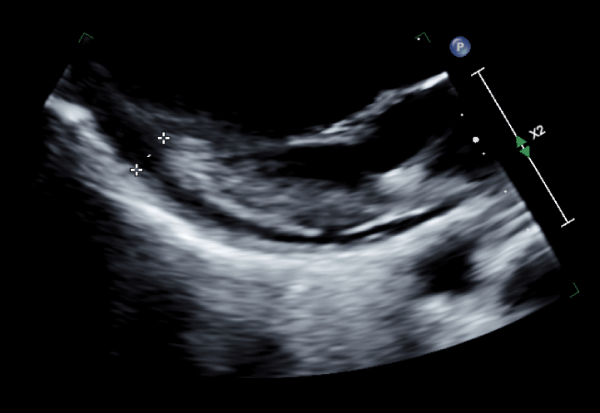 Figure 5: Marked pericardial effusion without Tamponade Noted on Echocardiogram.
View Figure 5
Figure 5: Marked pericardial effusion without Tamponade Noted on Echocardiogram.
View Figure 5
Her d-dimer was elevated (see Table 1) but workup was not remarkable for pulmonary embolism. Bilateral lower extremity ultrasound revealed no deep venous thrombosis, but did note significant bilateral inguinal lymphadenopathy.
Her overall acute infectious workup was negative, which prompted antibiotic and antiviral discontinuation. However, EBV serology suggested a past infection (see Table 1). At this point, the patient was given ketorolac with resolution of her pleuritic chest pain. Despite this, the patient continued experiencing recurring fevers and night sweats. Given her persistent symptoms, negative workup and elevated ferritin levels, hemophagocytic lymphohistiocytosis (HLH) was suspected as a differential diagnosis. Patient was given a dose of intravenous steroids with improvement of her symptoms and discharged home with urgent immunology and rheumatology follow up.
After the patient was initially suspected of developing symptoms concerning for pericarditis, she was treated with scheduled ibuprofen 400 mg every 8 hours and ketorolac 10 mg as needed for breakthrough pain, which significantly improved the pleuritic chest pain and discomfort. As her hospital course progressed and HLH or MAS was suspected, the patient received one dose of IV methylprednisolone 1g with a plan for long term outpatient taper with oral prednisone. Soon after discharge from the hospital, the patient was seen by rheumatology where she was started on a long-term steroid course with a plan to initiate immunomodulator therapy with anakinra.
CXCL9 and IL18 labs ordered and sent out. Inflammatory markers returned elevated (see Table 2) and were confirmatory for AOSD with secondary HLH. The patient was then started on steroids with a plan to start outpatient immunomodulator therapy with anakinra. Her symptoms were subjectively improving at discharge.
Table 2: HLH Serology. View Table 2
Initially, an infectious etiology was pursued when the patient was first admitted. As her hospital course progressed, however, an acute bacterial and fungal workup was unremarkable. Her clinical presentation thus far included fever, arthralgia, salmon‐colored rash, lymphadenopathy on imaging, negative ANA, negative rheumatoid factor. At that point, the primary care team believed the patient may have been experiencing an acute viral illness, which was complicated by a viral pericarditis presentation. The patient developed non ischemic chest pain, new EKG changes, and a new pericardial effusion, which satisfied 2 of 4 diagnostic criteria for acute pericarditis [4]. However, this did not explain the quotidian fevers, relapsing and remitting rash, elevated ferritin, or diffuse lymphadenopathy. When the patient’s clinical presentation and the EBV titers suggesting a past infection were taken into consideration, HLH was considered. Patients responded well with systemic steroids and confirmatory labs were drawn, which returned positive after the patient was discharged from the hospital.
The patient had an urgent outpatient rheumatology follow up scheduled where she satisfied the Yamaguchi Criteria for AOSD (see Table 3). At this point the patient was diagnosed with AOSD, but HLH was not strongly considered yet. Soon thereafter, the CXCL9 and IL-18 serologies resulted with marked elevations compared to the reference ranges consistent with MAS and Secondary HLH (see Table 2 and Table 4). The patient’s clinical course suggested a Secondary HLH diagnosis also due to a past EBV infection supported by her elevated serology [2]. Ultimately, the patient was finally diagnosed with the rare condition of Adult-Onset Still's Disease with Secondary Hemophagocytic Lymphohistiocytosis and started treatment.
Table 3: Yamaguchi Criteria for AOSD Diagnosis.
For the diagnosis of adult-onset Still disease, ≥ 5 criteria must be met, including ≥ 2 major [8]. View Table 3
Table 4: HLH- 2004 diagnostic criteria.
Diagnosis of HLH can be established if Criterion 1 or 2 is fulfilled [14]. View Table 4
Adult-Onset Still's Disease (AOSD) is an autoimmune condition that can trigger HLH in patients, amongst other severe inflammatory processes. Although described in the existing literature, this coincidence is rare [5]. Herein, a young female patient was diagnosed with Adult-Onset Still's Disease (AOSD) with Secondary HLH.
Hemophagocytic Lymphohistiocytosis (HLH) and Macrophage Activation Syndrome (MAS) can be severe consequences of several inflammatory disease processes like infection, cancer, or rheumatic diseases. HLH is a hyper inflammatory syndrome associated with high morbidity and mortality, but it has several variations: Primary HLH has a familial pattern and Secondary HLH and MAS are reactive [5,6]. However, there is a rare but identifiable association between AOSD complicated by Secondary HLH or MAS, which must be considered when assessing patients. Although rare, autoimmune conditions and viral infections can act as the catalysts for MAS and Secondary HLH, respectively [2]. In fact, some cases have described AOSD triggering HLH development [6,7].
Although secondary HLH is reactive to viral illnesses, there have been cases of HLH developing after vaccinations. Specifically, HLH after COVID-19 vaccination has been cited in the existing literature [8]. It is unknown if the patient in this case had received any recent vaccinations prior to her hospitalization, but her laboratory results were consistent with an already known viral association to Secondary HLH.
AOSD is a rare condition that can be diagnosed using the Yamaguchi Criteria. Given the rarity, diagnosing AOSD is challenging because there are no specific diagnostic tests. However, the Yamaguchi criteria has the highest sensitivity and includes Major and minor criteria. The major criteria include fever lasting one week, arthralgia lasting at least 2 weeks, salmon colored rash, and leukocytosis with granulocytosis. The minor criteria include sore throat, lymphadenopathy, hepatomegaly/splenomegaly, abnormal liver function tests, and negative results for ANA and RF. Yamaguchi, et al. 1992 also established exclusion criteria in the Journal of Rheumatology for diagnosing AOSD, which includes infections, malignancy, and other autoimmune processes [9]. This case seeks to describe an unusual case of AOSD perhaps triggered by a nonspecific viral illness. Therefore, AOSD should remain a possible differential diagnosis in patients who meet both Major and Minor Yamaguchi criteria despite meeting one or more exclusion criteria.
Previous studies have suggested that patients diagnosed with HLH secondary to AOSD have a worse prognosis and relapse rate [10]. The mortality of HLH ranges widely but still depends on the underlying cause with estimates of 2-20% mortality from rheumatic disease and 70% from certain lymphomas [11]. A major concern brought on by this case was a timely detection and diagnosis of HLH given its rarity and its dangerous associations. Like previously mentioned, HLH causes a severe hyper inflammatory process that damages organs, requiring ICU treatment. Unfortunately, HLH is nearly always fatal when discovered in the ICU. In a large retrospective cohort study of critically ill patients, ICU-HLH in adults was associated with a 57% mortality rate, regardless of HLH etiology or specific treatment [12]. Therefore, early detection and treatment are necessary for improved quality of life.
Despite nonspecific viral symptoms including sore throat, fever, myalgias, and arthralgias, it is important to identify AOSD with secondary HLH to improve long term quality of life with early management [5]. In the setting with characteristic physical exam findings, symptoms, profoundly elevated inflammatory markers, and confirmatory testing, clinicians must suspect AOSD and/or HLH. The HLH-2004 diagnostic criteria are the gold standard for HLH diagnosis [13]. However, multiple biomarkers should be considered to assist in diagnosing HLH in adults. Specifically, IFN-γ associated cytokines like IL18 and CXCL9 can help differentiate familial HLH from secondary HLH. Once a diagnosis is made, patients can receive treatment.
Controlling excessive inflammation and replacing the defective immune system [14]. The HLH-94 treatment protocol includes treatment with etoposide for the first phase and an allogeneic hematopoietic stem cell transplantation for the second phase to maintain remission. AOSD is treated using non-steroidal anti-inflammatory drugs (NSAIDs), steroids, disease modifying anti-rheumatic drugs (DMARDs), and other biologics. DMARDS used to treat AOSD include methotrexate, cyclosporine, leflunomide, azathioprine, mycophenolate, and cyclophosphamide. Biologic therapy with anakinra and tocilizumab have been gaining popularity in treating AOSD. Anakinra is an IL-1 blocker and is used to treat AOSD in patients who developed MAS/Secondary HLH. Second line biologic is Tocilizumab, which is an IL-6 blocker. Ultimately, patients may require a DMARD and biologic combination therapy for full control.
Hemophagocytic Lymphohistiocytosis (HLH) and Macrophage Activation Syndrome (MAS) can be a severe consequence of several inflammatory disease processes like infection, cancer, or rheumatic diseases. However, there is a rare but identifiable association between Adult-Onset Still’s Disease complicated by Secondary HLH or MAS, which must be considered. Although rare, autoimmune conditions and viral infections can act as the catalysts for MAS and Secondary HLH, respectively.
Adult Onset Stills Disease should remain a possible differential diagnosis in patients who meet both Major and Minor Yamaguchi criteria despite meeting one or more exclusion criteria including infections (sepsis and infectious mononucleosis), malignancy, or other rheumatic diseases (rheumatoid arthritis and/or polyarteritis nodosa) [8].
Previous studies have suggested that patients diagnosed with HLH secondary to AOSD have a worse prognosis and relapse rate [9]. Therefore, early detection and treatment are necessary for improved quality of life.
The HLH-2004 diagnostic criteria are the gold standard for HLH diagnosis [12]. However, multiple biomarkers should be considered to assist in diagnosing HLH in adults. Specifically, IFN-γ associated cytokines like IL18 and CXCL9 can help differentiate familial HLH from secondary HLH.
After ensuring the patient’s identity and protected health information will remain private, the patient graciously allowed us to share her story. However, she did not wish to make additional statements for the purpose of this publication.