Langerhans Cell Histiocytosis (LCH) is a rare disorder with an incident of 1.8 cases per 1,000,000. It involves granulomatous deposits in multiple organs, leading to a wide variety of manifestations such as bone lesions, pulmonary nodules, pituitary lesions, and skin lesions. With mucocutaneous manifestation, it is often diagnosed in childhood, typically making LCH a childhood disease. However, due to the involvement of multiple organ systems and variable clinical courses, if not diagnosed during childhood, the patients with LCH often suffer from delayed diagnosis and treatments as adults.
A 34-year-old Hispanic male with a medical history significant for insulin-dependent type 2 diabetes, recently diagnosed 1 month ago, presented with generalized weakness, left-sided rib pain, poor oral intake, polyuria, confusion and blurry vision. Initial laboratory studies revealed a sodium of 169, systolic blood pressure of 80 mmHg, urine osmolality of 77 mOsm/kg, and a serum osmolality of 358 mOsm/kg, consistent with diabetes insipidus (DI). Regarding the change in his mental status, a CT of the head without contrast was obtained. Enhancing on postcontrast imaging subsequently revealed an infiltrating lesion within the hypothalamus measuring approximately 2.1 × 1.5 × 1.9 cm (Figure 1). The lesion involved the proximal infundibulum without distal thickening. And the lesion was found to be widening the optic tracts and displacing the optic chiasm anteriorly. In addition, the posterior pituitary bright spot was not identified, suggestive of multiple pathologic processes including lymphocytic hypophysitis, granulomatous inflammation such as sarcoidosis, and neoplastic process such as astrocytoma, choroid glioma, and metastatic lesion. Given the concerning changes involving the pituitary as shown on radiographic findings, serum levels of the following pituitary hormones were obtained: FSH, LH, ACTH stimulation test, TSH, prolactin, IGF-1. The results came back with abnormally low FSH at 0.4 mIU/mL (reference range for male is 0.9 - 11.9 mIU/mL), LH < 0.1 mIU/mL (1.5-9.3 mIU/mL for male), ACTH stimulation test with a peak cortisol level at 14.8 ug/dL (normal response consists of at least 7 mcg/dL from baseline and a peak of 20 mcg/dL), TSH of 0.08 uIU/mL (0.35-4.94 uIU/mL), elevated prolactin 39.5 ng/mL (3.5-19.4 ng/mL), and low IGF-1 of 24 ng/mL (a z score of -3.3, below the reference range of -2.0 to + 2.0), thus revealing the presence of panhypopituitarism. In order to further delineate the nature of the pathologic process leading to panhypopituitarism, additional laboratory work-up was done including AFP tumor marker (normal at 5.2 ng/mL, reference < 6.1 ng/mL), B-hcg (normal at < 2 mIU/mL, reference < 5 mIU/mL), and ACE level to rule out sarcoidosis (normal at 34 U/L, reference 9 -67 U/L). In addition to the tumor marker work-up to look for potential primary source, CT of the chest, abdomen and pelvis with contrast was performed. This subsequently revealed a 10 × 17 mm lytic-appearing lesion on the left ninth rib, which matched the location of his complaint for rib pain. On the right sacral ala, there was a 1 cm lesion with sclerotic margins and multiple subcentimeter lesions on the left sacral ala. Additional findings included several thin-walled cysts throughout the lungs with a 4 mm pulmonary nodule in the right upper lobe (Figure 2). This subsequently led to an MRI of the pelvis without contrast. This revealed multiple bilateral lesions within the pelvis and left proximal femur, with the largest lesion in the left posterior ilium measuring 2.8 × 1.9 cm, with its appearance suggestive of bony metastatic disease. Biopsy was indicated, and a CT-guided biopsy of iliac bone was performed 3 days later. The biopsy results revealed the presence of rare atypical cells along with fibrosis, but without evidence of malignancy. Subsequently, the patient underwent biopsy of a suprasellar mass with a pathologic finding consistent with Langerhans Cell Histiocytosis with a mixed infiltrate containing Langerhans cells positive for CD1a, Langerin, S100, and BRAF V600E immunostain. A decision was made at neuro-oncology conference for him to receive approximately 20 Gy of radiation therapy to his hypothalamic mass, followed by a systemic chemo regimen with vemurafenib. The course of his radiation therapy spanned over a 40-day inpatient stay, as it was complicated by his short term memory deficits, waxing and waning mental status, and his inability to stay still for the duration of the radiation therapy necessary for him to receive the full dose. Following his admission which resulted in a biopsy-proven diagnosis and a chemo-radiation therapy, the patient suffered from frequent readmissions to the hospital (7 admissions within a 6 month-period) due to saddle pulmonary embolus, subsequent use of anticoagulation, and an anterior inter-hemispheric intracranial bleeding, resulting in an IVC filter placement, hypernatremia, hyponatremia along with repeated neurocognitive disorders with worsening confusion, generalized weakness, aggressiveness, and gait instability.
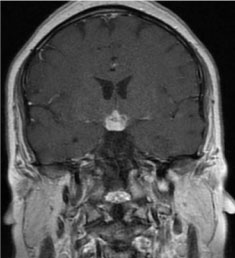 Figure 1: A 2.1 × 1.5 × 1.9 cm infiltrating hypothalamic lesion enhances avidly on postcontrast imaging with extension along the proximal infundibulum (MRI of head and pituitary with and without contrast, coronal section).
View Figure 1
Figure 1: A 2.1 × 1.5 × 1.9 cm infiltrating hypothalamic lesion enhances avidly on postcontrast imaging with extension along the proximal infundibulum (MRI of head and pituitary with and without contrast, coronal section).
View Figure 1
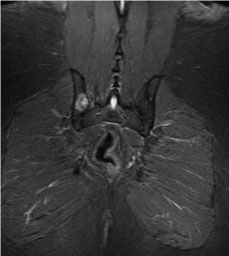 Figure 2: A lytic lesion noted on right pelvis (MRI of pelvis without contrast, coronal section).
View Figure 2
Figure 2: A lytic lesion noted on right pelvis (MRI of pelvis without contrast, coronal section).
View Figure 2
Langerhans cell histiocytosis (LCH) is characterized by abnormal proliferation of eosinophils, macrophages, giant cells, and lymphocytes. Historically, as the multiple different names suggest, LCH was considered a pathologic inflammatory process of the immune system [1]. With a discovery in 2010 revealing that half of the LCH cases were positive for BRAF V600E mutation, our understanding of the disease process shifted towards viewing LCH as a neoplasm of myeloid origin [2]. LCH is a childhood disease, with incidence of 8.9 cases per 1,000,000 in children under 15-years-old, with the median age of 3 at the time of diagnosis [3], due to its rather obvious mucocutaneous manifestation. Diagnosis of LCH in adulthood is even rarer, with incident of 1.8 cases per 1,000,000, with the median age at diagnosis being 33-years-old [4]. Epidemiologic studies in US pediatric cases revealed higher incidence of LCH among males, Hispanics, and patients with unfavorable socioeconomic background [5]. Although this case involves a 34-year-old adult male, he has many of the risk factors discussed above, as he is an un-documented, unemployed, Hispanic male speaking only Spanish. Owing to the rarity of the disease, along with various multisystem manifestations, by the time the patients are diagnosed with LCH, they have often already suffered from structural changes, especially with osseous lesions, and hypothalamic masses. One study revealed the bone as the most commonly affected system in patients with LCH (80% of all patients), while another study revealed DI as the most commonly presenting CNS symptoms (25% of all patients) [6,7]. Upon overcoming the diagnostic challenges stemming from its rarity and non-specific symptoms, multiple therapeutic challenges were addressed in this patient case, as there were no standardized therapies to guide treatment of LCH. This particular patient was amenable to vemurafenib treatment, as the histological findings revealed a BRAF V600E mutation. The decision to utilize vemurafenib was based on a previously published study of 14 patients resulting in a 41% response rate [2]. Despite seemingly favorable circumstances given his BRAF mutation, unfortunately existing literature also notes that patients with BRAF mutation are at an increased risk of neuro degenerative changes and treatment failure [8]. One of the challenges that the patient and his family faced include fluctuating mental status and difficulty in osmoregulation. These consist of behavioral changes such as aggressiveness and forgetfulness which lead to poor medication adherence necessary in treating his multiple endocrinopathies. Additionally, due to hypothalamic involvement leading to impaired thirst mechanism and osmoregulation, the patient suffered from significant changes in his serum osmolality and sodium level, often going from 160 mEq of sodium to 122 mEq during the same admission. To this day, the patient suffers from frequent re-admissions, spending no more than 2 weeks at a time at home between each admission. His current regimen includes desmopressin 0.2 mg bid, hydrocortisone 20 mg in the morning, and 10 mg in the evening, insulin, and 100 mcg of levothyroxine and vemurafenib to help manage his endocrinopathies. Due to the infiltrative process leading to the destruction of hypothalamic-pituitary tissues, it is believed that he will suffer from such endocrinopathies permanently. His prognosis is very poor, as one study noted of 50-66% mortality rate in those with multisystem LCH involvement [9]. All internists need to be aware of the myriad manifestations of this disease entity as this could potentially help in early detection and management of LCH. This would reduce the cost and morbidity associated with LCH (Figure 3).
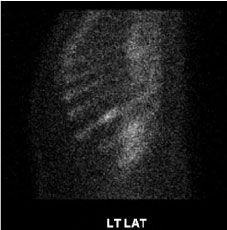 Figure 3: Abnormal left 9th rib corresponding to lytic defects seen on prior CT (Bone scan whole body, sagittal section).
View Figure 3
Figure 3: Abnormal left 9th rib corresponding to lytic defects seen on prior CT (Bone scan whole body, sagittal section).
View Figure 3