Cushing syndrome (CS) is a rare disease that results from prolonged supraphysiologic tissue action of glucocorticoids. This can be caused by a tumor of the adrenal glands, the lungs or the pituitary gland. When a pituitary tumor produces too much ACTH (adrenocorticotropic hormone), it causes the overproduction of cortisol by the adrenal glands. When the pituitary is the source of the over production, it is called Cushing's disease. Cushing's disease (CD) remains a diagnostic and therapeutic challenge. CD is the most frequent cause of endogenous CS and is associated with increased morbidity and mortality. Disease onset and severity reflects the magnitude of cortisol excess. Morbidity and mortality are increased in CD even in patients successfully treated and in remission. Despite early success in over 80% of the patients, in the long term CD recurs in almost 50% of the patients. The case is noteworthy for the atypical clinical presentation, severity and refractoriness of the hypokalemia. The biochemical testing has limitations, and these should be kept in mind in the investigation of hypercortisolism.
Cushing syndrome, Adrenocorticotropic hormone, Cushing disease, Pituitary, Hypokalemia, Cortisol
Hypercortisolism is a common condition in everyday clinical practice, but most cases are either obvious or not immediately relevant by themselves Cushing disease (CD) is the most frequent cause of endogenous Cushing syndrome (CS). It is characterized by an autonomous secretion of cortisol secondary to an underlying pituitary adenoma. Pituitary adenomas are thought to affect 10.6% of the population, mostly undiagnosed and asymptomatic, and among these, up to 13.8% may be adrenocorticotropic hormone (ACTH)-secreting [1,2]. Rarely does it occur in patients with multiple endocrine neoplasia type 1 or 4, familial isolated pituitary adenoma, McCune-Albright syndrome or Carney complex [3-5].
Cushing’s disease (CD) with an estimated incidence of around 1.5 per million per year, female preponderance (3:1) and peaking at ages between 25 and 45 years accounts for most of the cases of ACTH-dependent CS [3,4,6]. Lesions smaller than 1 cm (microadenomas) are more frequent than invasive lesions at the time of diagnosis. Patients with CD often exhibit numerous clinical and laboratory abnormalities. The prolonged supraphysiologic tissue action of glucocorticoids is believed to underlie the majority of the consequences of CS. Common features of Cushing’s disease are weight gain, hypertension, diabetes, poor short term memory, irritability, excess hair growth (women), red ruddy face, extra fat around the neck, round face, fatigue, poor concentration, and menstrual irregularity in addition to muscle weakness. Some of the less common features include insomnia, recurrent infection, thin skin and stretch marks, easy bruising, depression, weak bones, acne, balding (women), hip and shoulder weakness, violaceous striae, hypokalemia, unexplained osteoporosis, diabetes mellitus, and swelling of feet/legs [7].
As glucocorticoids are major counter regulatory hormones in carbohydrate metabolism, glucose intolerance and diabetes mellitus are, as expected, common features, affecting up to 50% of CS patients. Diabetes screening by means of fasting blood glucose levels fails to diagnose a significant portion of patients that have predominant postprandial hyperglycemia. Furthermore, hypokalemic metabolic alkalosis is not so frequent in CS, being present in up to 15% of cases [8]. Although CD remains the most frequent cause of endogenous hypercortisolism, acute, severe clinical presentations are more typical of ectopic ACTH secretion. This presentation may be explained by the fact that disease onset and severity reflect on how high and how fast cortisol levels have risen; in ectopic ACTH secretion, cortisol secretion is far more extreme than in any other CS subtype [9]. The authors report on a case of a patient that presented with severe hyperglycemia and refractory hypokalemic metabolic alkalosis that upon investigation was compatible with CD. In the case reports that have described a similar presenting clinical picture, the underlying cause was always ectopic ACTH secretion [10-13]. The diagnosis of CD was unexpected; therefore, we believe that this case report is noteworthy.
Not all patients have all of the signs and symptoms of Cushing’s disease. The diagnosis for Cushing’s disease is made by laboratory testing, which demonstrates the consistent overproduction of cortisol. The tests most commonly used are midnight salivary cortisol test, a 1 mg dexamethasone suppression test, or a 24h urinary free cortisol level [14,15]. All of these tests are approximately 92% accurate (2015, personal communication by Dr. Mary Lee Vance). A pituitary protocol magnetic resonance imaging (MRI) is also done to see if there is a visible tumor, if the blood ACTH level is detectable or elevated. Approximately 50% of patients with Cushing’s disease do not have tumors which are visible on MRI [7,15]. If no obvious tumor is seen, patients may have the inferior petrosal sinus sampling test. If the pituitary is the source of the excess cortisol production, the patient should undergo surgery by an experienced pituitary neurosurgeon. Cushing’s disease is characterized by a delay in diagnosis because of overlaps with other more common medical problems such as polycystic ovarian syndrome, diabetes, obesity and high blood pressure. The average time from onset of symptoms to diagnosis is 3-5 years [15].
A 35-year-old lady with a past medical history of obesity, diabetes mellitus (for 1 year- poorly controlled on pre mixed insulin and metformin), hypertension (for 2 years- well controlled on olmesartan and amlodipine combination) and glaucoma(for 1 year- on latanoprost eye drop), was admitted to the emergency department with the complaints of generalized weakness for last ten days. She had history of LUCS 3 months prior presentation. She also gained about 10 kg weight in last three months period. Her weakness was sub acute in onset, involving both upper and lower limbs simultaneously, aggravated over time, became so severe that she was unable to move without assistance which progressed to the point that she was completely bed ridden for 1 day. She had no history of diarrhea, vomiting, fever, neck pain, trauma to the neck, altered consciousness, sphincteric disturbance, speech or swallowing difficulty. She also denied history of taking heavy carbohydrate meals prior to the onset of symptom or any recent immunization.
On further inquiry, patient was having constant, dull aching, global headache with visual disturbance and episodic vomiting for the last 1.5 years for which she sought ophthalmologic consultation and diagnosed as a case of glaucoma and is still on treatment with little improvement. The patient emphatically insisted she was always very tired and that her body shape was changing with complains about hirsutism or generalized acne. There were no previous attempted pregnancies. There was no family record for endocrine diseases in direct relatives.
On admission, physical examination was remarkable for normal blood pressure, obesity (body mass index 31.2 kg/m), moon face, acne over face and upper chest, hirsutism (Figure 1), echymoses on the upper limbs (Figure 2). There was no peripheral edema, striae. Neurological examination revealed patient was conscious, oriented with normal speech and memory. Muscle power in all four limbs was MRC grade 2 with diminished all deep tendon reflexes with intact sensation. Fundoscopy revealed bilateral secondary optic atrophy (Figure 3) and visual field examination revealed bitemporal hemianopia.
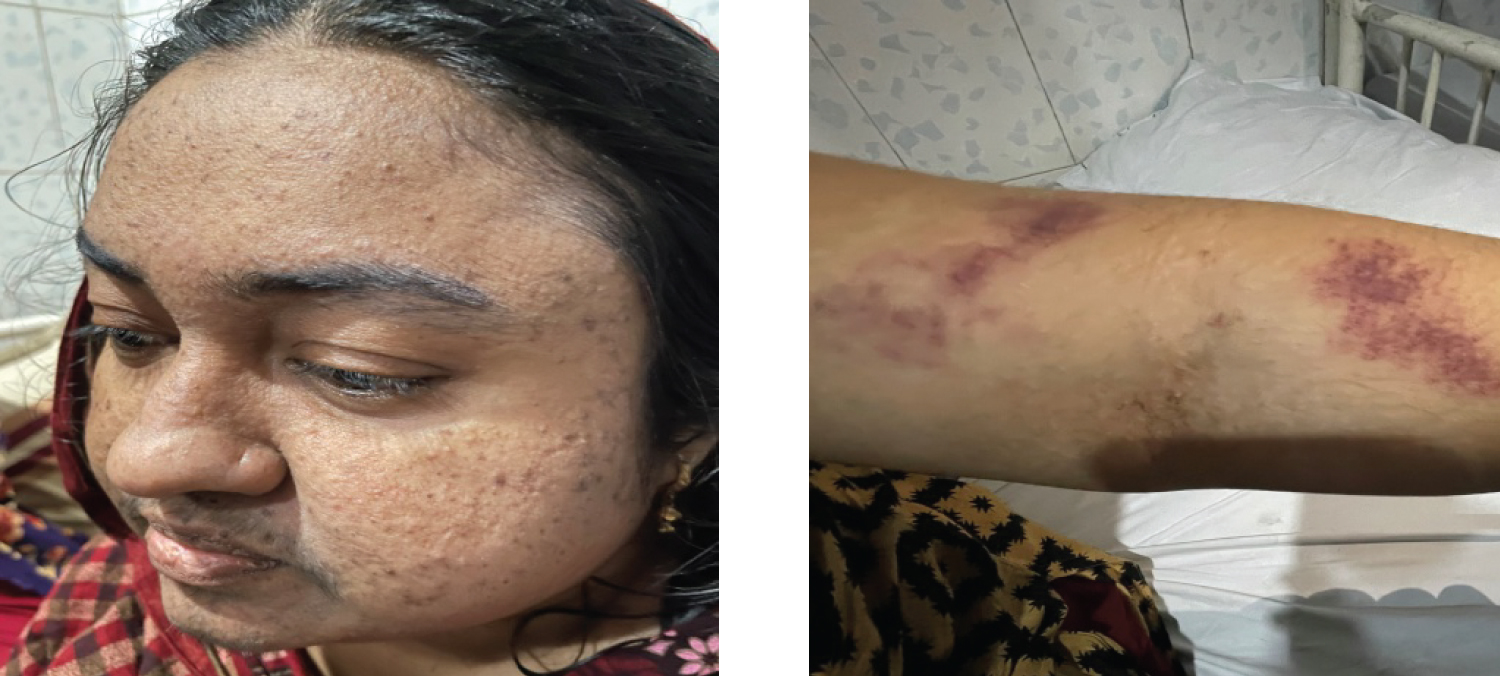 Figure 1 and 2: Moon face, acne, hirsutism over face and ecchymoses over left forearm respectively.
View Figure 1 and 2
Figure 1 and 2: Moon face, acne, hirsutism over face and ecchymoses over left forearm respectively.
View Figure 1 and 2
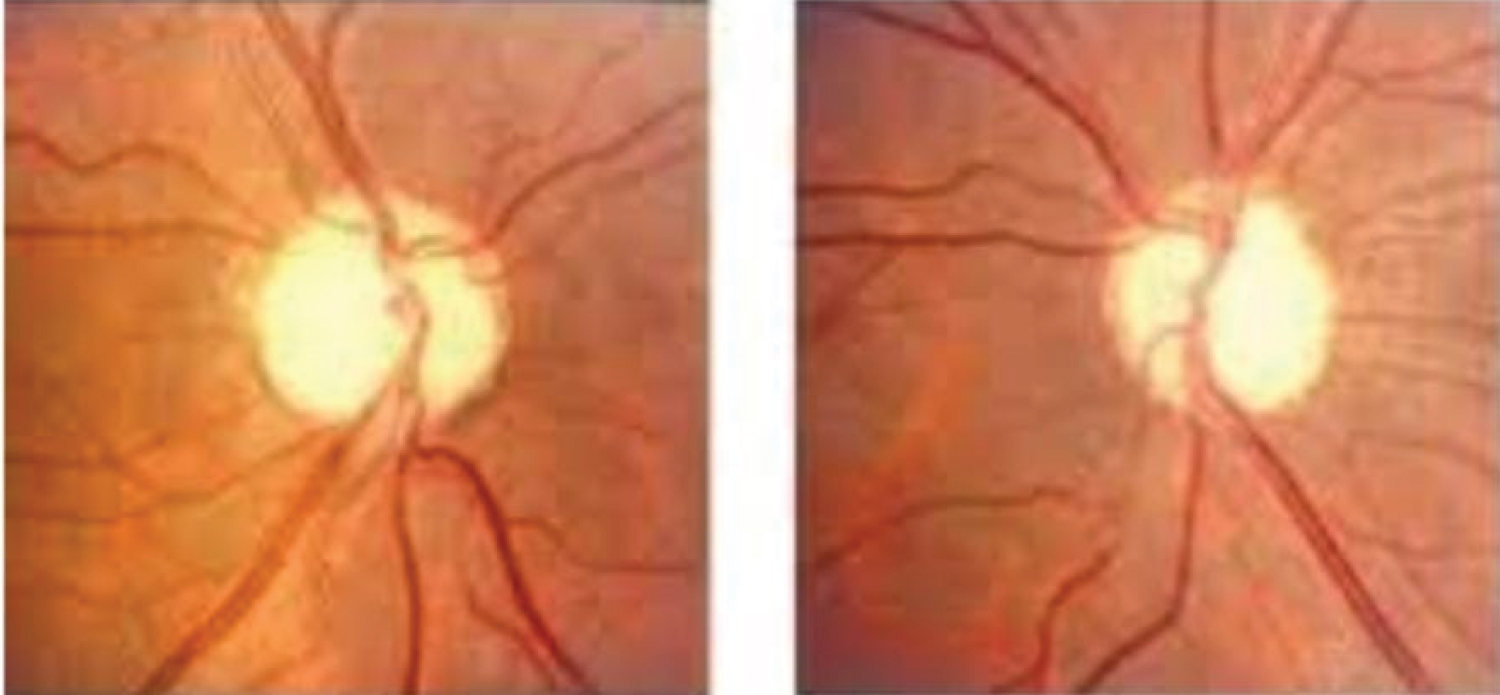 Figure 3: Fundoscopy revealed bilaretal secondary optic atrophy.
View Figure 3
Figure 3: Fundoscopy revealed bilaretal secondary optic atrophy.
View Figure 3
At admission, plasma glucose was 30.2 mmol/L, serum potassium 1.7 mmol/L, arterial pH 7.52, and bicarbonate 32 mmol/L, without evidence of underlying infection (unremarkable complete blood count and C-reactive protein levels). Electrocardiogram revealed ST depression, flattening of T wave and U wave in V2, V3, V4. Serum calcium and magnesium was normal. Urinary potassium was raised to 90 mmol/L. Basal cortisol (before 9 am) revealed raised cortisol 62 mcg/dl (normal 1.7-14.1 mcg/dl). TSH was supressed to 0.127 µU/L (normal 0.5-5.5 µU/L). Prolactin level was normal 5.4 ng/ml (normal 1.9-25.9 ng/ml) DHEA (dehydroepiandostenedione) and serum ACTH level was raised to 831 µgm/dl (normal 45-270 µgm/dl) and 352.1 pg/ml (normal 8.3-57.1 pg/ml) respectively. USG of whole abdomen revealed normal both ovaries and adrenal gland. She was started with both enteral and parenteral potassium suppliment with intensive insulin therapy. Despite treatment her potassium was 2.2 mmol/L on day two. High dose (8 mg) overnight dexamethasone suppression test was done which revealed suppressed morning basal corsol level suggestive of cushing disease. (Baseline cortisol at 11:00 PM- 68.9 ug/dL Morning cortisol at 9:00AM (the following day)- 61 ug/dL). Perimetry revealed bitemporal hemianopia (Figure 4 and Figure 5). MRI of brain was undertaken which showed pituitary macroadenoma with suprasellar and parasellar extension (Figure 6).
 Figure 4 and 5: Perimetry revealed bitemporal hemianopia.
View Figure 4 and 5
Figure 4 and 5: Perimetry revealed bitemporal hemianopia.
View Figure 4 and 5
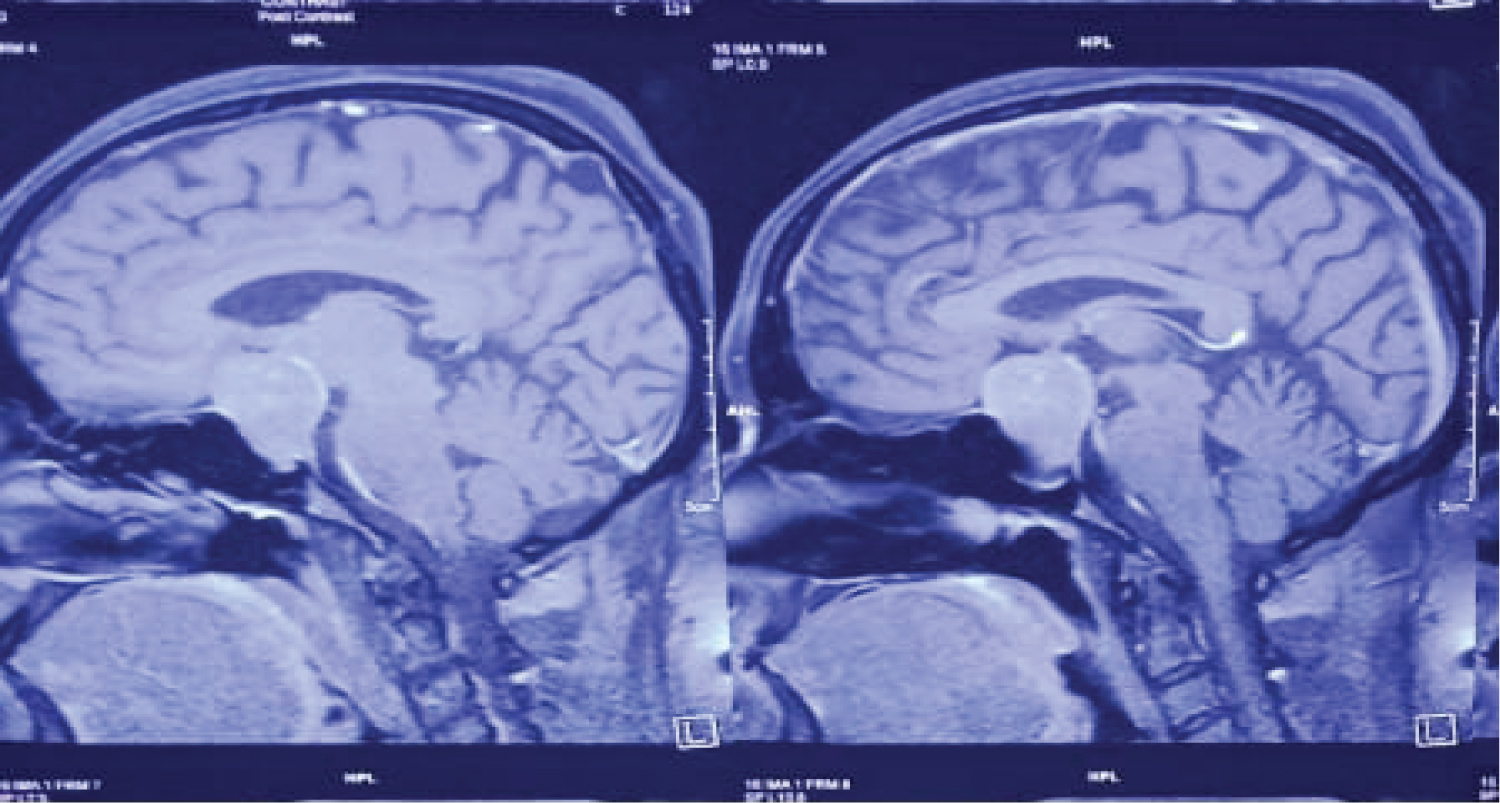 Figure 6: MRI of brain revealed pituitary macroadenoma with parasellar and suprasellar extension.
View Figure 6
Figure 6: MRI of brain revealed pituitary macroadenoma with parasellar and suprasellar extension.
View Figure 6
So, final diagnosis of resistant hypokalemia due to cushing disease with steroid induced glucoma with hypertension and diabetes was made. She was started perioprative treatment with ketoconazole.She was referred to higher center for neurosurgical management.
Cushing’s is a challenging disease to diagnose. The diagnosis is often delayed because Cushing’s is frequently masked by its overlap with more common medical problems such as diabetes, high blood pressure, obesity, and polycystic ovary syndrome. Cushing’s may be more common than previously thought.
CS is characterized by a constellation of signs and symptoms that develop due to excessive, prolonged tissue exposure to inappropriate high glucocorticoid levels. The majority of cases are attributable to exogenous corticosteroid use (iatrogenic CS). Endogenous Cushing is far less common, with an estimated incidence of 0.7 to 2.4 per million populations. It may be etiologically divided into ACTH-dependent (80 to 85% of cases) and ACTH independent disease [16].
CD accounts for 75 to 80% of ACTH-dependent CS cases, is more prevalent in females, and has the highest incidence in the fourth decade. The source of excessive circulating cortisol is usually an ACTH-secreting pituitary microadenoma (< 1 cm). Ectopic ACTH syndrome (EAS) is less common, being responsible for 15 to 20% of endogenous CS cases. It is more frequent in males and occurs later than CD. Excessive ACTH tends to be produced by an extrapituitary tumor such as pulmonary (45% of all EAS cases: bronchial carcinoid, pulmonary small cell or adenocarcinoma), thymic (11%), and gastrointestinal neuroendocrine tumors (5%), islet cell tumors (8%), medullary thyroid carcinomas (6%), and pheochromocytomas (5%). Epithelial neoplasms may also be associated with EAS; however, they are far less frequent [9]. Adrenal CS, on the other hand, is the most frequent cause of CS in the pediatric age group. Rare causes of CS include corticotrophin releasing hormone-secreting neoplasms and cyclic CS [16].
Clinical presentation of CS will vary according to gender, age, underlying cause, duration and severity of hypercortisolism, and the rapidity of biochemical glucocorticoid rise. Men usually present at a younger age with a more severe clinical manifestations, such as violaceous striae, muscle weakness, osteoporosis, and urolithiasis [16]. Obesity, hypogonadism, and psychiatric and cognitive dysfunction are highly prevalent in both genders [16]. Metabolic complications are also common, namely, hypertension in 70 to 80%, glucose intolerance in 45 to 70%, hypercholesterolemia in 70%, and osteoporosis in 50 to 80%. Hypokalemic metabolic alkalosis affects mostly EAS patients (90% vs. 10 to 15% in CD) [16]. According to etiology, circulating cortisol levels are usually higher in patients with EAS, explaining the rapid onset (over 1 month) and severity of the clinical picture in these cases. EAS secondary to more indolent tumors may be clinically indistinguishable from CD, in which the classical clinical and metabolic abnormalities of CS are of gradual onset (over months to years) [12]. Diagnosis relies on confirmation of autonomous cortisol secretion with different tests: 1 mg and low-dose dexamethasone suppression test, midnight salivary cortisol, 24-hour urinary free cortisol, and cortisol rhythm. After establishing the diagnosis, establishing the cause is the next step and includes ACTH measurement. If ACTH is elevated, additional tests are needed to distinguish CD from EAS (8 mg dexamethasone suppression test [overnight or 48-hour] and inferior petrosal sinus sampling), and according to the findings, cranial MRI or nuclear imaging (Octreoscan, Ga-DOTATATE), respectively, may be warranted. ACTH levels below 5 pg/mL warrant adrenal imaging, and if needed, genetic testing (Carney complex, PRKAR1A, ARM5) [13].
New diagnostic approaches to the differential diagnosis of CS emphasize the central role of the CRH or desmopressin test [17]. In the CRH test, a 30-40% increase in ACTH and a 20% increase in cortisol are generally used to define CD; while in ectopic CS baseline ACTH and cortisol values are generally much higher and the response is much less; and in chronic CRH over secretion a decreased ACTH response and increased cortisol response are described. By these criteria, in this case the CRH test suggests CD albeit imperfectly, and is strongly against the other possibilities [4,7,17-22].
Common pitfalls in the diagnosis of CD do not seem to explain discrepancies; free cortisol is increased as evaluated by the 24-h urinary cortisol excluding cortisol-binding globulin (CBG) increase induced by pill; no drugs were used that could increase the metabolism of dexamethasone or present anti-glucocorticoid effects [4,7,17-19].
Imagiological findings are also discordant. In classic cases a small (5 mm) round hypodense (T1-weighted sections) lesion is found; it may be hyperdense in T2-weighted sections and may not uptake the contrast [3,4,7,23,24]. Most interestingly is the pituitary stalk widening; this finding has been reported in relation to granulomatous or immune-mediated pituitary diseases in regard with hypopituitary states [3,4]. Given the global involvement of the pituitary it may instead in this case correspond to the enlargement of the pars tuberalis that surrounds the pituitary stalk.
First-line therapy is almost always surgical. Transsphenoidal surgery in CD is associated with relapse in 20% of patients, while adrenal causes are often curable, with an excellent prognosis in benign disease. EAS, on the other hand, is difficult to manage, since most of the ACTH-secreting neoplasms are already metastasized at the time of diagnosis and may require either medical therapy or bilateral adrenalectomy. Corticosteroid cover is required during and for some time after surgery, since long-term hypercortisolism suppresses both nonadenoma corticotrophes (ACTH-dependent cases) and the “healthy” adrenal (unilateral ACTH-independent causes). This long term suppression leads to a temporary adrenal ‘stunning’ that requires time to fully functional recovery. Physiologic glucocorticoid replacement may be required for more than 1 year after surgery. Pituitary radiotherapy may be an option if there is residual tumor after surgery. Its effects are delayed for years and usually require medical therapy until the radiotherapy has an effect. Medical therapy is only indicated as pre-operative preparation in patients with contra-indications to surgery, relapsed and occult disease cases, and as a bridge to the effects of radiotherapy. Pharmacologic options include ketoconazole, metyrapone, mitotane, etomidate, mifepristone, pasireotide, and cabergoline. Most of the agents have a poor side effect profile, and since some are adrenolytic, may warrant concomitant glucocorticoid supplementation [16].
The reported case illustrates an acute, severe presentation of CS, which would usually be suggestive of an ectopic ACTH source; however, the absence of an identifiable extrapituitary tumor along with the presence of a pituitary macroadenoma led to a clinical dilemma. On the one hand, the adenoma appeared to be invasive and was the probable cause the hypopituitarism, therefore favoring a surgical indication. On the other hand, incidental pituitary adenomas are present in 10.6% of the population, and the macroadenoma could have been just an incidentaloma, if not for the concomitant hypopituitarism, since the clinical and biochemical picture were both highly suggestive of EAS. From this case, it is clear that pituitary adenomas can also give rise to florid CS presentations.
The reason behind the acute presentation is not clear. The large tumor size, along with the low mitotic index, could suggest that the adenoma was not rapidly growing. No typical clinical data of long-term ‘subclinical’ Cushing suggest a previously undiagnosed CD that suddenly progressed. The tumor was probably an inefficient secreting neoplasm that for unknown reasons suddenly became hormonally active. The authors speculate that beyond tumor mass and mitotic index, other mechanisms might be at play, namely, pro opiomelanocortin processing and/or ACTH packaging defects. A few case reports (mostly affecting women before the seventh decade) have described silent macroadenoma progression to florid CS; however, clinical evidence data are still scarce for this seemingly distinct clinical entity [25].
After establishing the diagnosis of CS, the etiologic diagnostic strategies also have some limitations. The 8 mg dexamethasone suppression test relies on the principle that pituitary adenoma ACTH secretion is sensitive to prolonged extremely high doses of exogenous glucocorticoids. However, the failure to suppress cortisol secretion after high-dose dexamethasone (to less than 50% of baseline cortisol concentration) has been shown to have poor accuracy, sensitivity, and specificity (76.5, 79.3, and 66.7%, respectively) in the differentiation between CD and EAS. Furthermore, pituitary ACTH-secreting macroadenomas, compared with microadenomas, are often less suppressible in dynamic testing, possibly explaining some of the unexpected biochemical results [26]. Inferior petrosal sinus sampling also has limitations, most of which are of technical character. This approach requires the expertise of a high-volume center, as the imprecise placement of the central catheters may be a significant source of error [27].
Cushing’s is difficult to diagnose and increases morbidity and mortality in patients who are untreated. CD often presents with the gradual onset of a constellation of signs and symptoms of prolonged supraphysiologic cortisol levels. Sudden and dramatic onset of CS often suggests an ectopic ACTH-secreting tumor. These cases are more frequently accompanied by altered mental status, hyperglycemic emergencies, and hypokalemia. The described case illustrates an atypical presentation of a severe case of CD with new, marked hyperglycemia and severe refractory hypokalemia. It is noteworthy to emphasize the importance in the suspicion of a secondary cause of diabetes mellitus in the presence of hyperglycemia emergencies. Furthermore, the biochemical investigation of ACTH-dependent CS is complex and subject to inaccuracy, justifying a holistic approach and goal-oriented therapy in all cases.
None declared.