Amyotrophic Lateral Sclerosis (ALS) or Lou Gehrig's disease is a neurodegenerative and neuromuscular disorder characterized by a progressive death of motor neurons & motor paralysis that culminates in death, usually within 3-5 years of diagnosis from respiratory failure due to paralysis. Currently approved ALS therapies are not curative and fail to increase healthy lifespan. The worldwide prevalence of ALS is expected to increase by 69% over the next 25 years, yet its etiology remains scientifically unverified, complicating the discovery and development of effective therapies. Through this cross-sectional clinical study employing microbiological and cellular analyses of fecal and blood samples isolated from human patients with and without ALS, we achieved to confirm recently emerging pre-clinical and clinical evidence linking ALS with intestinal dysbiosis & systemic microbial infection, and thus provide rationale for future scientific investigations of intestinal dysbiosis as a potential therapeutic target for improving and extending the healthy lifespan of human patients diagnosed with ALS.
Amyotrophic Lateral Sclerosis (ALS) or Lou Gehrig's disease is a neurodegenerative disorder characterized by progressive degeneration of both upper and lower motor neurons, and manifests as progressive physical paralysis that culminates in death (most often due to respiratory failure) within an average of 3-5 years from disease onset [1-3]. ALS is the most common adult-onset motor neuron disease (MND), with a worldwide annual incidence of about 2 per 100,000 [4]. The worldwide prevalence of this fatal disease is expected to increase by up to 69% over the next 25 years [5]. ALS affects individuals of all races and ethnicities (both male and female); however, whites, males, non-Hispanics, individuals aged > 50 years, and those with a family history of ALS are more likely to develop this disease [6-8]. There is currently no cure for ALS, and its etiology remains elusive. The only 2 drugs approved by the FDA to treat this disease are riluzole (a glutamate inhibitor) and edaravone (an anti-oxidant), both of which fail to halt the progression of this disease and fail to consistently prolong healthy lifespan (healthspan) of ALS patients beyond several months. Thus, there is an urgent need for the ethical scientific discovery, development, and delivery of therapeutics that consistently extends the healthy lifespan (healthspan) of ALS patients beyond what is currently possible. The etiology of ALS has not been fully elucidated in the peer-reviewed scientific literature; however, evidence is emerging to indicate that disturbances in the intestinal microbiota are involved in the cause & progression of ALS and other neurodegenerative diseases [9]. Here, we provide clinical evidence linking intestinal dysbiosis & systemic microbial infection with ALS and discuss rationale for further scientific investigation of this link and it's potential as a therapeutic target.
Data from the blood and fecal analyses of 82 human patients were used in this retrospective cross-sectional study. The Steenblock Research Institute (SRI) & its Ethics Committee approved of this retrospective cross-sectional clinical study. All human patients were > 18 years of age & were able to provide informed consent. Of the 82 human patients included in this study, 54 (26 male & 28 female) were diagnosed with ALS per the revised El Escorial criteria, and 28 (14 male & 14 female) did not have ALS or ALS-related symptoms and served as a control group in this study. At the time of biological (blood and feces) sample collection & analyses, the mean age of human patients with ALS (ALS Patients) was 63.5 ± 12.3 years, and those without ALS (Non-ALS Patients) was 59.4 ± 13.4 years.
Concentration of fecal secretory-IgA was determined by an enzyme-linked immunosorbent assay (ELISA). Serum white blood cell differential was determined by automated cell counting. Screening for a panel of 5 intestinal pathogenic bacteria was conducted per culture-based microbiological methodology. LabCorp, Doctor's Data, and Great Palms Laboratory completed all afore mentioned fecal & blood samples analyses per contemporary best practices in clinical laboratory science.
Differences between groups (ALS Patients vs. Non-ALS Patients) were determined by ANOVA with Student's t-test. Data are expressed as mean ± standard error of the mean (SEM). A P < 0.05 was considered as statistical significance.
 Figure 1: Mean concentration of fecal secretory-IgA is consistently & significantly higher in ALS relative to Non-ALS Patients (323.96 ± 30.73 mg/dL vs. 106.71 ± 5.56 mg/dL; P < 0.001).
View Figure 1
Figure 1: Mean concentration of fecal secretory-IgA is consistently & significantly higher in ALS relative to Non-ALS Patients (323.96 ± 30.73 mg/dL vs. 106.71 ± 5.56 mg/dL; P < 0.001).
View Figure 1
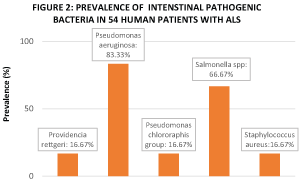 Figure 2: Fecal carriage of one or more bacterial species was detected in 100% of the 54 Human ALS Patients in this study.
View Figure 2
Figure 2: Fecal carriage of one or more bacterial species was detected in 100% of the 54 Human ALS Patients in this study.
View Figure 2
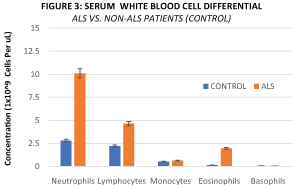 Figure 3: Mean serum concentration of Neutrophils (10.09 × 109 Cells vs. 2.78 × 109 Cells, P = 0.036), Lymphocytes (4.63 ×109 Cells vs. 2.19 × 109 Cells, P = 0.015), and Eosinophils (1.95 × 109 Cells vs. 0.14 × 109 Cells, P = 0.023) are significantly higher in ALS relative to Non-ALS Human Patients.
View Figure 3
Figure 3: Mean serum concentration of Neutrophils (10.09 × 109 Cells vs. 2.78 × 109 Cells, P = 0.036), Lymphocytes (4.63 ×109 Cells vs. 2.19 × 109 Cells, P = 0.015), and Eosinophils (1.95 × 109 Cells vs. 0.14 × 109 Cells, P = 0.023) are significantly higher in ALS relative to Non-ALS Human Patients.
View Figure 3
Intestinal dysbiosis is widely defined as an abnormal elevation in the concentration of pathogenic microbial species that reside within the intestinal lumen and has recently been associated with the etiology and progression of many neurodegenerative diseases [9]. Secretory immunoglobulin A (SIgA), an endogenous antimicrobial factor measurable in feces, is naturally found in the external secretions bathing mucosal surfaces of the intestine at a concentration that is positively proportional to the concentration of pathogenic microbial species that reside within the intestinal lumen [10]. This cross-sectional clinical study found that the mean concentration of fecal secretory immunoglobulin A (SIgA) is significantly higher (> 300%) in ALS Patients relative to Non-ALS Patients (Figure 1), thereby indicating that ALS Patients harbor a significantly higher concentration of pathogenic microbial species in their intestinal lumen and are thus in a state of intestinal dysbiosis. Furthermore, fecal carriage prevalence of one or more pathogenic bacterial species was confirmed in 100% of ALS Patients enrolled in this study (Figure 2). Among healthy human populations, fecal carriage prevalence of the pathogenic bacteria pseudomonas aeruginosa and salmonella spp is 8.2% and 2-3%, respectively [11-13]. However, in this study's population of ALS Patients, fecal carriage prevalence of pseudomonas aeruginosa and salmonella spp is found to be 83.3% and 66.7%, respectively (Figure 2), thereby indicating an increased susceptibility in the ALS Patients' intestine to harbor pathogenic bacterial species relative to that of healthy non-clinical populations. This study's finding of intestinal dysbiosis as a feature in ALS is in agreement with smaller cross-sectional clinical studies conducted in 2016 [14] and 2017 [15], both of which evaluated the intestinal microbiome of ALS Patients through different methodologies to confer status of intestinal dysbiosis.
This study's analysis of serum white blood cell concentration indicates that human ALS Patients have a significantly elevated neutrophil-to-lymphocyte ratio (Figure 3), which is an indication of systemic inflammation in response to systemic microbial infection [16,17]. Intestinal dysbiosis has been shown to cause intestinal barrier dysfunction or intestinal epithelial hyperpermeability in a mouse model of ALS [18,19], thereby providing an anatomical pathway for the migration of pathogenic intestinal microbes and/or their metabolic byproducts into the systemic circulation via the gut circulatory system to elicit systemic microbial infection. Given the co-occurrence of intestinal dysbiosis and systemic microbial infection in 100% of the ALS Patients enrolled in this study, it is plausible that a similar physiological phenomenon is occurring in human ALS Patients. This study's finding of systemic microbial infection as a feature of ALS Patients is consistent with that of a previous smaller study reporting the presence of serum pathogenic microbial byproducts & systemic inflammation in human ALS Patients [20].
In a recent pre-clinical study utilizing a mouse model of ALS, it was demonstrated that intestinal dysbiosis precedes the development and progression of ALS symptoms, and that reversal of intestinal dysbiosis with a dietary probiotic intervention alleviates ALS symptoms and progression, reverses intestinal barrier dysfunction, and prolongs healthy lifespan [19]. This strongly indicates that intestinal dysbiosis may be a modifiable cause of ALS. Through this cross-sectional clinical study, we achieved to confirm recently emerging pre-clinical and clinical evidence linking ALS with intestinal dysbiosis & systemic microbial infection, and thus provide rationale for future scientific investigation of intestinal dysbiosis as a potential therapeutic target for extending the healthy lifespan of human patients diagnosed with ALS.
T.I. and D.A.S. performed data collection & patient examinations. T.I. performed data analysis, prepared the figures, & wrote the manuscript. M.J.A composed & revised the manuscript per reviewer feedback. A.S.S.A. & E.W. revised the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. Authors claim no conflicts of interest related to the research described in the manuscript.