DNA aptamers were developed against human growth hormone releasing peptide (GHRP)-6 and the major metabolite of GHRP-2 (pralmorelin) known as AA-3 (D-Ala-D(β-naphthyl)-Ala-Ala-OH) in 10% human serum or 50% human urine. The lead 5'-biotinylated candidate aptamers from ELISA-like microplate screening were conjugated to commercially available Dynal™ streptavidin-polystyrene-coated 2.8 µm diameter magnetic (magnetite) microbeads (MBs) and used to "pull down" and purify or enrich for their cognate targets from buffer as well as undiluted human serum and urine. Aptamer binding was detectable at low ng levels in buffer, but not in serum or urine by the ELISA-like (ELASA) assay. Similarly, aptamer-MB pull down was detectable in buffer by electrophoresis in Coomassie blue-stained 20% polyacrylamide gels, but gel detection in serum and urine was compromised. In two cases, lead GHRP-6 candidate aptamers were shown to pull down an interfering protein in the vicinity of 50 kD from serum by electrophoresis. AA-3 and GHRP-6 were pulled down using aptamer-coated MBs and detected in buffer, serum and urine by mass spectrometry (MS) in 81.25% (13 of 16 trials), thus attesting to the potential of aptamers for use in the detection of doping with these peptides in athletes. The 18.75% (3 of 16 trials) negative detection results by MS for aptamer-MB pull down trials were rectified when 5X more aptamer-MB reagents were added or when the aptamer-MBs were used in the body fluid matrices in which they were selected (i.e., the aptamers were placed in their intended chemical environments). The aptamer-coated MB pull down method is generalizable to enrichment and sensitive detection of other analytes in serum and urine as well using aptamers selected in these body fluid matrices.
Aptamer, GHRP, Growth hormone, Magnetic bead, Peptide, Pull down
Athletes who engage in doping for performance enhancement have become increasingly sophisticated in their strategies to avoid detection. Thus, it is no longer adequate to simply test for recombinant human growth hormone (hGH) or its induced longer-lived effector IGF-1 [1,2]. Some athletes are currently doping with a family of growth hormone releasing peptides (GHRPs) known to induce secretion of hGH (secretagogues) [3] which are delineated in Barroso, et al. [4]. Although, liquid chromatography-mass spectrometry (LC-MS) and immunoassay techniques already exist for detection and quantification of these GHRP and related peptide hormones [5-8], antibody development is well known to be hindered by a lack of immunogenicity by low molecular weight (MW) peptides which require conjugation to carrier proteins for antibody development. Nucleic acid aptamers are developed entirely in vitro and obviate the need for host animals or even cell culture [9]. In addition, some antibodies are known to exhibit imperfect batch-to-bath reproducibility [9-11] which may affect inter-laboratory reproducibility for testing labs. Aptamers do not suffer from lot-to-lot variations in composition, because their known DNA sequences can be precisely reproduced time and again by chemical synthesizers [9].
Thus, after consultation with GHRP experts at the World Anti-Doping Agency (WADA) about the most important GHRP targets, the authors sought to develop a set of high affinity and highly specific DNA aptamers to detect the major metabolite of pralmorelin or GHRP-2 known as AA-3 and GHRP-6 (abbreviated G6 herein) whose structures and MWs are shown in Figure 1, because they had developed successful high-affinity and highly specific aptamers to several other physiologically active peptides previously [12,13]. The authors further sought to demonstrate the utility of the lead GHRP aptamers when attached to magnetic microbeads (MBs) to pull down, purify and enrich [14,15] their cognate analytes from buffer, human serum and urine with validation by gel electrophoresis and mass spectrometry (MS) as reported herein.
 Figure 1: Structures and molecular weights of the target peptides. View Figure 1
Figure 1: Structures and molecular weights of the target peptides. View Figure 1
AA-3 (D-Ala-D(β-naphthyl)-Ala-Ala-OH) and GHRP-6 (His-D-Trp-Ala-Trp-D-Phe-Lys-NH2) were both synthesized by Genway Biotech (San Diego, CA) and purified to greater than 98.0% as assessed by reverse-phase HPLC. The peptides were dissolved and diluted up to 500 µg/mL in sterile phosphate buffered saline (PBS, pH 7.2) conjugated to Dynal™ (Life Technologies, Inc., Carlsbad, CA) tosyl-M280 (2.8 µm diameter) MBs at a ratio of 40 µg of peptide per 100 µL of stock tosyl-MBs by virtue of having primary amines in their structures. Nine rounds of MB-SELEX DNA aptamer development were conducted according to previously published protocols [1,2,12,13] except that the aptamers were either selected in 10% pooled human AB serum (Mediatech Inc., Cat. No. 35-060-CI, diluted in sterile nuclease-free water) or 50% human urine (pooled from one of the author's families) to emulate the chemical environments in which the aptamers were intended to bind. After each round of SELEX, the presence of a 100 bp aptamer PCR product in a 2% agarose gel stained with ethidium bromide was used to verify the SELEX process and enable progression to the next round. All DNA SELEX templates, primers as previously described [1,2,12,13], and biotinylated candidate aptamers were synthesized at Integrated DNA Technologies, Inc. (IDT; Coralville, IA).
Round 9 GHRP aptamers were cloned into chemically competent E. coli using a GC (high Guanine-Cytosine content) cloning kit from Lucigen Corp. (Middleton, WI) and sequenced at Sequetech Inc. (Mountain View, CA) using proprietary high GC DNA sequencing techniques. The total of 142 unique individual DNA aptamers which resulted for the two GHRP targets (69 aptamer sequences for AA-3 and 73 sequences for GHRP-6) were synthesized as separate 5'-biotinylated oligonucleotide sequences and lyophilized at 4 nanomoles per well in individual wells of 96-well plates and purchased from IDT. Secondary stem-loop structures of the top four aptamers from Table 1 were determined by entering the individual DNA sequences into free internet UNAFold software on IDT's website (https://www.idtdna.com/UNAFold) using 25 ℃ and 145 mM sodium as parameters.
Table 1: DNA sequences of the lead anti-GHRP aptamer candidates. View Table 1
To determine relative affinity rankings for each of the candidate GHRP aptamer sequences, an ELASA [1,2,13] was conducted by first immobilizing 250 ng per well of the GHRP peptides in 100 µL of 0.1 M NaHCO3 (pH 8.5) overnight at 4 ℃ in covered, flat-bottom, polystyrene 96-well plates (Greiner Bio-One GmbH, Frickenhausen, Germany; Cat. No. 655101). Plates were decanted and washed three times in 200 µL of sterile PBS plus 0.001% Tween 20 (PBST). Wells were then blocked with 150 µL of 2% ethanolamine in 0.1 M NaHCO3 for 1 h at 37 ℃ and decanted, followed by three more washes with 200 µL of PBST. The unique 69 AA-3 and 73 G6 5'-biotinylated candidate aptamer DNA sequences from the round 9 pool (Table 1) were then added to their respective wells at 500 picomoles per well in 100 µL of PBS and mixed at room temperature (RT; ~25 ℃) on a rotary mixer for 1 h. Wells were decanted and washed three times in 200 µL of PBST for at least 5 min per wash with gentle mixing. One hundred µL of a 1:3,000 dilution of streptavidin-peroxidase (SAv-HRP; Thermo-Fisher, Waltham, MA; Product No. 21126) from a 1 mg/mL stock solution was added per well for 30 min at RT with gentle mixing. Wells were again decanted and washed three times with 200 µL of PBST as before. One hundred µl of One-Component® ABTS substrate (Kirkegaard Perry Laboratories, Gaithersburg, MD), which had been equilibrated to RT, was added per well for 15 min at RT, and reactions were halted by addition of 100 µL of 1% SDS. Absorbance was quantified using a microplate reader at 405 nm.
Identical ELASA technique was used for GHRP titration experiments except that 100 µL of AA-3 or G6 containing 1,000 ng/mL each peptide in 0.1 M NaHCO3 was serial two-fold diluted in 100 µL of 0.1 M NaHCO3 per well across a microtiter plate. No target was added to some wells to serve as background blanks and the 500 picomoles of each lead aptamer were added in 100 µL of PBS, undiluted human serum or human urine and 5'-biotinylated aptamers were allowed to bind for 1 h at RT with gentle mixing. Pooled and certified disease-free human blood serum and urine were purchased from Bioreclamation IVT, LLC (Hicksville, NY) for the ELASA titration and aptamer-MB pull down studies.
One hundred µl (160 mg) or 500 µL (800 mg) of stock Dynal™ streptavidin (SAv)-polystyrene-coated M280 2.8 µm diameter MBs (Thermo-Fisher Corp.) to be used per sample were washed twice in 1 mL of sterile phosphate buffered saline (PBS, pH 7.2) for 5 min per wash on a Dynal™ MPC® magnetic rack [14,15]. All four lead 5'-biotinylated DNA aptamers from Integrated DNA Technologies, Inc. (Coralville, Iowa, USA) were rehydrated at ~2.2 mg/ml in sterile PBS and 100 µL of aptamers were added per 100 µL of stock SAv-MBs and mixed at room temperature (RT) for 1 h in sterile 1.7 mL microfuge tubes. Aptamer-5'-biotin-SAv-MBs (hereby referred to as aptamer-MBs) were collected on the Dynal™ MPC® magnetic rack for 2 min and washed 3 times in 1 mL of sterile PBS per wash. Aptamer-MBs were resuspended in their original volume of sterile PBS, stored at 4 ℃ and then used in pull down assays as follows. In initial studies, 100 µL of stock aptamer-MBs were added per sample tube to 1 mL of sterile PBS, human serum or urine as indicated and spiked with 100 µg of each GHRP target. In subsequent studies, the level of aptamer-MBs was increased to 500 µL per sample tube with 100 µg of each GHRP per microfuge sample tube in PBS, serum or urine. Tubes were mixed gently end over end on a rotary mixer at RT for 2 h. Tubes were washed 3 times in 1 mL of sterile PBS per wash on the MPC® magnetic rack with MB resuspension in PBS following magnetic collection for 2 min. Captured GHRPs and other molecules were eluted off of the aptamer-MBs by resuspension of the aptamer-MBs in 50 µL of 0.1 M HCl for 5 min at RT. Aptamer-MBs were again collected for 5 min on the MPC® magnetic rack and the eluate was siphoned out with a pipette tip and transferred to a fresh microfuge tube. The pH of the eluate was neutralized with 50 µL of 0.1 M NaOH [15].
Twenty µL of each pull down sample were mixed with 20 µL of Invitrogen Novex® 5X TBE (Tris-Borate-EDTA) sample loading buffer and electrophoresed in various wells of 20% polyacrylamide TBE gels at 200 V in cold 1X TBE buffer for ~1 h followed by Coomassie Blue staining (Invitrogen Simply Blue™ stain) and/or Invitrogen SilverQuest™ silver staining according to the manufacturer's instructions. Gels were also loaded with 7-10 µL of ColorBurst™ (8-220 kD; Sigma Chemical Co., St. Louis, MO, Cat. No. C1992) or Spectra™ Multicolor Low Range (1.7-40 kD; Thermo-Fisher, Cat. No. 26628) MW markers for reference to the probable relative MWs of detected bands. The remaining 80 µL of each eluted and pH-neutralized sample were sent on ice to the core mass spectrometry laboratory at the University of Texas Health Science Center at San Antonio (UTHSCSA) for electrospray ionization time-of-flight (ESI-TOF) mass spectrometry analysis by comparison with the > 98.0% purified standard AA-3 and G6 stock peptide samples.
Table 1 reveals the DNA sequences of the top four lead aptamer candidates as determined by extensive ELASA screening and ranking of the highest affinity (darkest green or highest Absorbance at 405 nm) candidates from among the 142 total aptamer candidates for AA-3 and G6 aptamers (ELASA screening data not shown for brevity). From Table 1, it appears that runs of multiple cytosines (C) flanked by one or more thymines (T) in three of the lead candidates excluding the constant 18-base PCR primer regions (italicized) on the 5' and 3' ends was a common finding, although other shared sequence runs between the candidates appeared to be present as well. The notion that some of these high C and T content regions could be binding regions for the GHRPs was supported by the secondary structural analyses generated by UNAFold shown in Figure 2 where runs of multiple Cs flanked by Ts are noted in the boxed regions.
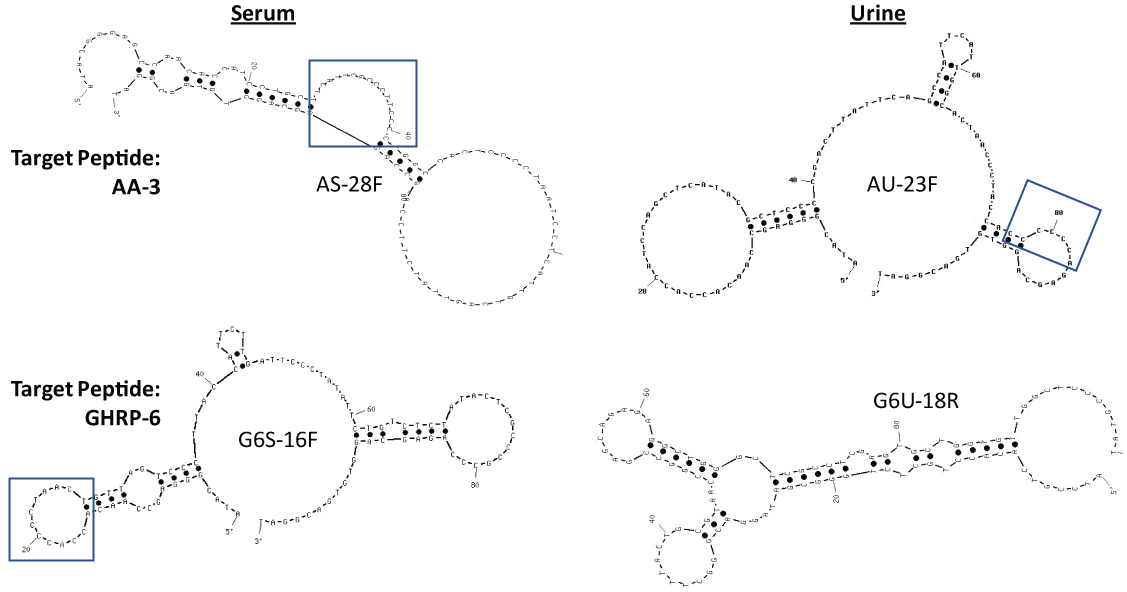 Figure 2: Secondary stem-loop structures of the lead GHRP aptamers generated by use of free internet UNAFold software using 145 mM Na+ and 25 ℃. The highest affinity (lowest ∆G) structures are shown in each case. Boxed regions indicate C- and T-rich regions from Table 1 which also form loop regions in the 2-dimensional structures and may be favorable areas for GHRP binding. View Figure 2
Figure 2: Secondary stem-loop structures of the lead GHRP aptamers generated by use of free internet UNAFold software using 145 mM Na+ and 25 ℃. The highest affinity (lowest ∆G) structures are shown in each case. Boxed regions indicate C- and T-rich regions from Table 1 which also form loop regions in the 2-dimensional structures and may be favorable areas for GHRP binding. View Figure 2
Figure 3 illustrates that while the G6 aptamers developed in serum (G6S-16F) and urine (G6U-18R) in their designations) appeared to have significantly higher affinity than the AA-3 aptamers (AS-28F and AU-23F), three of the four lead aptamers appeared to have limits of detection in the mid to low nanogram per well ranges (i.e., remained above the mean background level indicated by the dotted line) when tested by ELASA in PBS. All four titration curves completely flattened out in human serum and urine (body fluid data not shown because it was flat in all cases). This failure to detect AA-3 or GHRP-6 in serum and urine by ELASA could be due to much larger proteins from the body fluid samples adhering to and obscuring the immobilized small peptides (AA-3 or G6) on the surface of the wells, but this failure may be specific to the ELASA surface detection methodology and appeared to be overcome by the use of aptamer-coated magnetic beads and pull down methodology [14,15].
 Figure 3: Results of different titration trials for the direct AA-3 and G6 enzyme-linked sandwich assays (ELASAs) using the lead aptamers from Table 1 or Figure 2 in PBS. Means and standard deviation error bars for three independent trials (N = 3) are shown. The dotted line indicates the baseline mean for the background absorbance at 405 nm level without aptamers. Results from human serum and urine ELASA titrations (not shown) were flat perhaps due to larger proteins adhering to the wells and preventing aptamer binding. View Figure 3
Figure 3: Results of different titration trials for the direct AA-3 and G6 enzyme-linked sandwich assays (ELASAs) using the lead aptamers from Table 1 or Figure 2 in PBS. Means and standard deviation error bars for three independent trials (N = 3) are shown. The dotted line indicates the baseline mean for the background absorbance at 405 nm level without aptamers. Results from human serum and urine ELASA titrations (not shown) were flat perhaps due to larger proteins adhering to the wells and preventing aptamer binding. View Figure 3
Figure 4 Illustrates that all four lead aptamers appeared to pull down their cognate target peptides in buffer (PBS) spiked with 100 µg of AA-3 or G6 when the biotinylated aptamers were bound to 100 µL of stock streptavidin-MBs (lanes 2-5) especially when compared to 2 µg standards of the AA-3 (lane 6) and G6 (lane 7) peptides (boxed region for lanes 2-7) after Coomassie blue staining. The gel in Figure 4 does contain some anomalies such as the diffuse band for G6 pulldown with G6U-18R in lane 5 and the higher ~10 kD bands in lanes 6 and 7 for the peptide standards as well as the smear in the boxed region of lane 7. Some of these anomalous results may be attributable to charge effects and aggregate formation due to the lack of sodium dodecyl sulfate (SDS) in the gel or samples to equalize all charges across the board. The authors wished to use SDS for more precise MW determinations. However, when SDS was added in other gel electrophoresis experiments (gels not shown), it appeared to eliminate detection of the putative peptides by Coomassie blue staining.
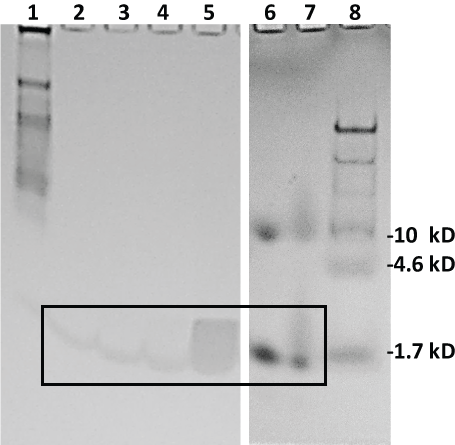 Figure 4: Coomassie blue-stained 20% polyacrylamide gel electrophoresis results from the aptamer-MB pull down conducted in buffer (PBS). Lane 1: 7 µL of 8-220 kD Sigma ColorBurst™ MW markers; lane 2: 20 µL of eluted pull down material from the AS-28F aptamer-MBs; lane 3: 20 µL of eluted pull down material from the AU-23F aptamer-MBs; lane 4: 20 µL of eluted pull down material from the G6S-16F aptamer-MBs; lane 5: 20 µL of eluted pull down material from the G6U-18R aptamer-MBs; lane 6: 2 µg of AA-3 standard peptide; lane 7: 2 µg of GHRP-6 (G6) standard peptide and lane 8: 10 µL of Thermo-Fisher Spectra™ Multicolor Low Range (1.7-40 kD) MW markers. Lanes 6-8 were run in a separate 20% polyacrylamide gel under identical conditions. The boxed region encloses faint, but detectable, bands suggesting detection of the AA-3 and G6 peptides. View Figure 4
Figure 4: Coomassie blue-stained 20% polyacrylamide gel electrophoresis results from the aptamer-MB pull down conducted in buffer (PBS). Lane 1: 7 µL of 8-220 kD Sigma ColorBurst™ MW markers; lane 2: 20 µL of eluted pull down material from the AS-28F aptamer-MBs; lane 3: 20 µL of eluted pull down material from the AU-23F aptamer-MBs; lane 4: 20 µL of eluted pull down material from the G6S-16F aptamer-MBs; lane 5: 20 µL of eluted pull down material from the G6U-18R aptamer-MBs; lane 6: 2 µg of AA-3 standard peptide; lane 7: 2 µg of GHRP-6 (G6) standard peptide and lane 8: 10 µL of Thermo-Fisher Spectra™ Multicolor Low Range (1.7-40 kD) MW markers. Lanes 6-8 were run in a separate 20% polyacrylamide gel under identical conditions. The boxed region encloses faint, but detectable, bands suggesting detection of the AA-3 and G6 peptides. View Figure 4
The gel image in Figure 5 illustrates the lack of detection of the very low MW peptide bands when the aptamer-MB pull down was conducted in human serum spiked with 100 µg of AA-3 or G6 and the 20% polyacrylamide gel was Coomassie blue stained. However, interestingly, two clear bands of a protein or proteins with apparent MW of ~50 kD were pulled down from human serum by the two G6 aptamers (lanes 4 and 5 of Figure 5).
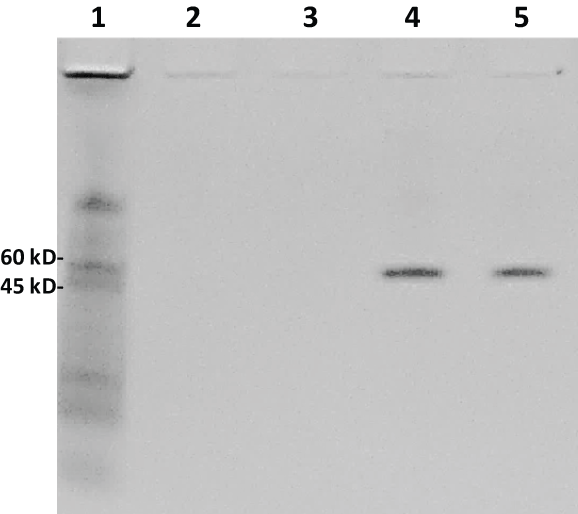 Figure 5: Another Coomassie blue-stained 20% polyacrylamide electrophoresis gel from the aptamer-MB pull down conducted in human serum. Lane 1: 7 µL of 8-220 kD Sigma ColorBurst™ MW markers; lane 2: 20 µl of eluted pull down material from the AS-28F aptamer-MBs; lane 3: 20 µL of eluted pull down material from the AU-23F aptamer-MBs; lane 4: 20 µL of eluted pull down material from the G6S-16F aptamer-MBs; lane 5: 20 µL of eluted pull down material from the G6U-18R aptamer-MBs. Although no low MW peptide bands are seen with Coomassie blue staining, clear ~50 kD protein bands were pulled down by the G6S-16F and G6U-18R aptamers (lanes 4 and 5) from human serum. View Figure 5
Figure 5: Another Coomassie blue-stained 20% polyacrylamide electrophoresis gel from the aptamer-MB pull down conducted in human serum. Lane 1: 7 µL of 8-220 kD Sigma ColorBurst™ MW markers; lane 2: 20 µl of eluted pull down material from the AS-28F aptamer-MBs; lane 3: 20 µL of eluted pull down material from the AU-23F aptamer-MBs; lane 4: 20 µL of eluted pull down material from the G6S-16F aptamer-MBs; lane 5: 20 µL of eluted pull down material from the G6U-18R aptamer-MBs. Although no low MW peptide bands are seen with Coomassie blue staining, clear ~50 kD protein bands were pulled down by the G6S-16F and G6U-18R aptamers (lanes 4 and 5) from human serum. View Figure 5
Staining of the same gel shown in Figure 5 with the more sensitive silver stain demonstrates that very low MW bands do appear in Figure 6A lanes 2-5 (boxed) which may correspond to AA-3 and G6 peptides. These same low MW bands, however, are not seen in the adjacent gel image (Figure 6B) from aptamer-MB pull down samples obtained from human urine spiked with 100 µg of AA-3 or G6. A number of higher MW bands do appear which derive from serum and urine when stained with the more sensitive silver stain indicating that trace levels of other proteins probably nonspecifically adhere to the aptamer-MBs.
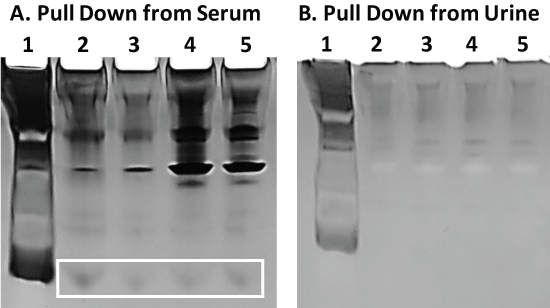 Figure 6: Panel A) The same gel shown in Figure 5 following fixation and silver staining. The boxed region may enclose the low MW AA-3 and G6 peptides which were not visible with the less sensitive Coomassie blue stain in Figure 5; Panel B) A similar 20% polyacrylamide gel electrophoresis result for the same aptamer-MB pull down assay scheme described and conducted in Figure 5 but in human urine instead of serum. No low MW peptide bands were observed for the pull down assay from urine despite the more sensitive silver staining. View Figure 6
Figure 6: Panel A) The same gel shown in Figure 5 following fixation and silver staining. The boxed region may enclose the low MW AA-3 and G6 peptides which were not visible with the less sensitive Coomassie blue stain in Figure 5; Panel B) A similar 20% polyacrylamide gel electrophoresis result for the same aptamer-MB pull down assay scheme described and conducted in Figure 5 but in human urine instead of serum. No low MW peptide bands were observed for the pull down assay from urine despite the more sensitive silver staining. View Figure 6
While electrophoresis of the supposed low MW peptides in Coomassie blue or silver stained polyacrylamide gels was suggestive of some successful aptamer-MB pull down, the ultimate test to validate pull down of the AA-3 and G6 was to detect and identify these peptides by MS in buffer, human serum and human urine. The results of this effort with the UTHSCSA's Mass Spectrometry Core Laboratory are summarized in Table 2 which gives the results of four separate trials using either 100 µL (Trial 1) or 500 µL (Trials 2-4) of aptamer-MBs in PBS, human serum or urine. In Table 2, the plus symbol represents positive detection at any signal intensity and the minus symbol represents the inability to detect a given target peptide by ESI-TOF MS, albeit the qualifier (low +) was used to indicate a barely perceptible G6 peptide peak in one case. The Table does not document the results of negative control studies in which streptavidin-MBs alone were added to 100 µg of AA-3 or G6 spiked into PBS, human serum or urine, but these negative controls did not produce any perceptible peptide peaks (i.e., the negative controls were negative).
Table 2: Summary of Aptamer-MB pull down experiments using mass spectrometry detection. View Table 2
Analysis of the three failures (-) and one dubious (low +) result in Table 2 reveals that they occurred when either the lesser 100 µL amount of aptamer-MBs was used or the aptamer used was not developed in the environment in which it was used. Thus, increasing to 500 µL of aptamer-MBs enabled G6S-16F to achieve positive detection of GHRP-6 (compare Trials 1 and 2). This was not the case for the G6U-18R aptamer when the level of aptamer-MBs was increased from 100 to 500 µL in PBS, in fact detection went from barely perceptible to negative (compare Trials 1 and 2 for G6U-18R). Detection using 500 µL of G6U-18R in serum also failed but changed to successful positive detection in urine (Trial 4 of Table 2 for G6U-18R). This observation strongly suggests that the G6U-18R aptamer must be in a higher urea concentration or at least in the urine milieu in order to fold into the correct conformation to bind and detect GHRP-6. Bruno observed similar chemical environmental effects on a cancer biomarker aptamer (ERK2) in previous work in which 2 mM EDTA was added to bind any free divalent cations (to mimic the diluted blood environment in which the aptamer) was selected. The removal of divalent cations was required to make the ERK2 aptamer fold properly, bind and pull down ERK2 [15]. No quantitative or semi-quantitative data were obtained by MS, because this would require costly studies, although one can surmise that detection levels of the AA-3 and G6 peptides were low since it is known that ESI-TOF MS can detect sub-ng levels of peptides [5,6].
The data presented herein represent a first attempt to develop DNA aptamers capable of binding and pulling down GHRPs in body fluids to detect athletes who are doping with these hGH secretagogues. The data are generally encouraging and suggest that optimization such as determining the exact optimal level of aptamer-MBs to add to a given sample volume should be the next step in assay development. While it is true that relatively large amounts of the target peptides (100 µg each) were spiked into the buffer, serum and urine samples, the surface area of aptamer-coated MBs is probably relatively small for capture of the abundant 100 µg/mL targets, thus leading to low levels of pulled down target materials. It is, however, certain that aptamer pull down is specific since the use of simple streptavidin-coated MBs without the tethered aptamers (negative controls) failed to detect AA-3 or G6 peptides by MS (data not shown). Interestingly, the two G6 aptamers (G6S-16F and G6U-18R) whether developed in diluted 10% serum (S) or 50% urine (U) clearly pulled down a protein from serum in the range of ~50 kD. Analysis using the University of Delaware's Protein Information Resource database and Peptide Match search engine [16] produced zero hits when the full GHRP-6 peptide sequence (HWAWFK) or truncated versions of the GHRP-6 sequence such as HWAWF or WAWFK were used as search terms. However, the partial sequence WAWF produced over 30 hits within the human proteome and one in particular for human heavy chain Fab antibody fragments (e.g., http://www.uniprot.org/uniprot/A2NYU6) which is very plausible to find in human serum and might weigh ~ 50 kD, if degraded, although numerous other serum proteins are possible interferents. This observation of competitive interference from serum proteins (bands in lanes 4 and 5 of Figure 5) while problematic for detection of GHRP-6 in serum, does also attest to the specificity of the G6 aptamers. And if the target for these G6 aptamers is truly WAWF, then the authors may be able to find other aptamers among their final pool of 73 candidate G6 aptamers that can bind the histidine (H) or lysine (K) ends of GHRP-6 (HWAWFK) for enhanced specificity in serum samples.
Finally, it is interesting to note from Table 2 that increasing the level of aptamer-MBs from 100 to 500 µL helped one of the mass spectral analyses for the G6S-16F aptamer to achieve detection (i.e., to become positive between Trials 1 and 2 in PBS), presumably due to the 5X increased surface area provided by five times more MBs. And it is interesting that the G6U-18R aptamer either failed or performed marginally in Table 2 unless it was placed in urine, the environment in which it was selected. This fact, strongly suggests that the aptamer folding can be influenced by high urea levels or other chemical factors such as divalent cation concentration as Bruno observed in a separate aptamer-MB pull down study [15]. These observations also underscore the importance of selecting and developing aptamers in a chemical milieu as close as possible to the environment or body fluid in which the aptamers are intended to be used.
While, the presently reported aptamers may never actually be used to detect athlete doping with GHRP-2 or -6, they illustrate the potential of aptamers to detect small peptides and other analytes in magnetic pull down assays where antibodies may fail due to the lack of peptide immunogenicity during development in vivo. In addition, aptamers can be developed faster and less expensively than antibodies without the use of animals or cell culture. Thus, aptamers are reagents that magnetic bead analyte separation techniques including those coupled to MS detection such as SISCAPA (Stable Isotope Standards and Capture by Anti-Peptide Antibodies) [17-20] may wish to consider along side or in lieu of antibodies for small analyte detection in body fluids.
This work was funded by World Anti-Doping Agency (WADA) Grant No. 16B02JB. The authors are grateful to the Core Mass Spectrometry Laboratory at the Univ. of Texas Health Sciences Center at San Antonio, especially Mr. Sam Pardo, for contracted mass spectrometry and consultation.