The purpose of the current study was to prepare Methotrexate (MTX) loaded Poly Lactic-Co-Glycolic Acid (PLGA) Nanoparticles (NPs) and investigates their toxicity effect on human glioblastoma cells. The influence of different experimental parameters including polymer concentration, Polyvinyl Alcohol (PVA) concentration in the external phase and drug concentration on the particle size was evaluated. Size of NPs determined by Dynamic Light Scattering (DLS) and Scanning Electron Microscopy (SEM) was from 150 to 200 nm. The Encapsulation Efficiency (EE) and Drug Loading (DL) corresponding to the optimal conditions were 62.96% and 5.72%, respectively.
Differential Scanning Calorimetric (DSC) and Fourier Transform Infrared Spectroscopy (FTIR) results indicated that MTX was dispersed as amorphous phase in the PLGA matrix and the chemical structure of MTX loaded PLGA did not change. Moreover, in vitro release profiles of MTX from NPs exhibited a sustained release behavior and the release rate of MTX from the NPs increased as the medium acidity enhanced. in vitro Cytotoxicity studies revealed that the cell Cytotoxicity effect of MTX loaded PLGA NPs on U87MG cells was more than free MTX. In conclusion, MTX loaded PLGA NPs could be considered as potential candidate for drug delivery in the treatment of glioblastoma.
Methotrexate, Poly (lactic-co-glycolic acid), Nanoparticles, Glioblastoma, Sustained release
Glioblastoma Multiforme (GBM) is considered as the most common and lethal primary brain tumor as well as the most malignant neoplasm of the astrocytic regions, accounting for 15% of all primary brain tumors, about 50-60% of all astrocytomas and 60 to 70% of malignant gliomas [1-3]. Despite advances in the treatment of GBM, the median survival of patients has not been changed significantly [4,5]. Numerous challenges and obstacles exist for the treatment of these tumors including: First; challenges related to the specific characteristics of GBM tumors such as uncontrolled cell growth, high aggressive power, tendency to recurrent micro-vascular growth, resistance to apoptosis and genetic instability [6] and second; challenges related to Blood-Brain Barrier (BBB), Blood-Cerebrospinal Fluid Barrier (BCB) and Blood-Tumor Barrier (BTB) [7,8] as well as third; challenges related to chemotherapeutic agents and the drug delivery to glioblastoma tumors such as insufficient permeability of physiological barriers, general toxicity and drug resistance [9]. However, many problems can be improved by delivery of sufficient amount of an appropriate chemotherapeutic agent to glioblastoma tumors using an suitable pharmaceutical carrier.
Nano-scale drug delivery systems have potential to target chemotherapeutic agents to tumor cells over a prolonged period of time, leads to increase in efficacy and decrease in toxic side effects [10]. Polymeric NPs are considered as optimal carriers for drug delivery due to their appropriate characteristics. One of the most polymers used in the field of nano-drug delivery systems is PLGA. PLGA, a synthetic copolymer is composed of glycolic and lactic acid units which are linked together by ester linkage, thus bringing aliphatic linear polyester [11]. Many applications and attentions to this polymer in drug delivery field is because of its properties such as formulations and techniques of the production conformed by various types of drugs (e.g. hydrophilic or hydrophobic molecules or macromolecules), biodegradability and biocompatibility, FDA approval in drug delivery systems for medical applications, protection of drug from degradation, possibility of sustained release, facility in modifying surface properties, resulting in better interaction with biological materials and facility in targeting NPs to specific points [12]. Therefore, PLGA NPs have potential to improve drug delivery systems and overcome many problems related to the treatment of brain tumors. MTX, a folic acid antagonist, is a hydrophilic small molecule (log P, -1.8, MW, 454.5 and poor aqueous solubility (0.01 mg/ml)) which is commonly used as clinical chemotherapy agent and highly efficacious antineoplastic drug [13,14]. MTX inhibits Dihydrofolate Reductase (DHFR) and interfere with tumor cell DNA, RNA and consequently protein synthesis, leads to inhibition of the proliferation of tumor cells [15]. However, using methotrexate has been restricted because of limitations such as low half-life in plasma, low penetration into the brain and dose-dependent systemic side effects [13-16]. To overcome this problem, MTX can be loaded in various NPs such as liposomes [17], solid lipid NPs [18], cationic bovine serum albumin [19], poly (butyl cyanoacrylate) [20], nanogels [21], dendrimers [22]. In several studies stated PLGA nanoparticles have ability to cross through BBB by numerous mechanisms [23-25]. MTX loaded into a suitable nano-carrier such as PLGA can improve the efficiency of the drug on glioblastoma tumors and also reduce adverse effects of MTX. In this study, MTX loaded PLGA NPs were prepared by emulsion solvent evaporation method and the effect of different parameters on the particle size was investigated. Moreover, encapsulation efficiency, drug loading, physical characteristics and drug release profile were studied. The cytotoxicity effects of MTX loaded PLGA on human glioblastoma U87MG cells was investigated by 3-(4,x5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay.
PLGA (lactide/glycolide = 50:50, Resomer® 503H) was purchased from Boehringer Ingelheim Pharma Gmbh & Co. Germany. MTX (purity > 97%, CasNo: 7413-34-5) was bought from Excella GmbH company. PVA with molecular weight 30,000-70,000, Phosphate-Buffered Saline (PBS) and tetrazolium dye 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide were from Sigma-Aldrich (USA). All other solvents and reagents were in analytical grade. Dulbecco's Modified Eagle's Medium (DMEM) and Fetal Bovine Serum (FBS) were purchased from Gibco® (USA).
MTX loaded PLGA NPs prepared by double-emulsion solvent evaporation method. MTX (5 mg) was dissolved in acetone (5 ml) and slowly added to the organic phase containing chloroform (5 ml) and PLGA (50 mg). The obtained solution was sonicated for 1 min. The polymeric solution was added drop wise to the aqueous phase containing PVA 0.1 w/v% (20 ml) under stirring at slow speed. The obtained emulsion was sonicated for 2-3 min in bath sonication. Afterwards, a solution of PVA 0.1 w/v% (10 ml) was added to the emulsion and placed on a magnetic stirrer with 800 rpm for 12 h. The NPs were separated by centrifugation at 15,000 g for 20 min and washed with distilled water. The suspension was freeze dried at -80 ℃ for 8 h and lyophilized by vacuum pressure less than 50 m Torr and at the temperature of -40 ℃ for 24 h and also the collected supernatant was analyzed for free drug. Blank PLGA NPs were prepared according to the same procedure. In order to identify the effect of various parameters on size distribution and encapsulation efficiency, the parameters of PVA, PLGA and MTX concentration were investigated by single factor experiment.
Particle size and polydispersity index of samples after adequate dilution were analyzed by a particle size analyzer (Malvern Instruments, model Zetasizer® 3000-HS, UK) instrument using DLS technique. Zeta Potential (ZP) data were performed through electrophoretic light scattering (20 ℃, 150 V), in triplicate for each sample (Malvern Zetasizer Nano-ZS, Malvern Instruments, UK).
The particle size and morphology of the NPs were studied by SEM (Philips, model XL30, The Netherlands). A suitable sample of NPs was mounted on metal stubs and the samples were coated by sputtering gold for 180 sec at 14 mA under argon atmosphere for secondary electron emissive SEM and then the morphology of the NPs was observed at an acceleration voltage of 15 kV.
For determination of MTX EE, NPs were separated from supernatant by centrifugation at 15000 g (Eppendorf centrifuge 5810R) for 20 min at 22 ℃. Free MTX concentrations were measured in the supernatants by UV-Vis spectroscopy (Optizeri 2120 UV plus) in λmax = 306 nm [26,27]. The concentration of free MTX in the supernatant was obtained by comparing the absorption of the supernatant to the standard curve related to absorption and MTX concentration. The drug EE (%) and DL (%) were calculated by the following equations:
The effects of drug encapsulation on the chemical group of the formed components and the interaction between the components were studied by FTIR (model Bruker Equinox 55). The FTIR spectra were collected at 20 ℃ from 500-4000 cm-1 for samples of MTX, PLGA copolymer and PLGA-MTX NPs.
DSC analysis of the samples was accomplished by DSC. The appropriate amount of the samples of MTX, PLGA copolymer, PLGA/MTX mixture and PLGA-MTX NPs were heated under inert nitrogen atmosphere on an aluminum pan at a heating rate of 10 ℃/min from 0 to 300 ℃.
The kinetics of MTX release from the optimized MTX loaded PLGA NPs was investigated in aqueous milieu. The 20 mg from prepared drug-loading NPs with optimized preparation method were dispersed in 10 ml PBS, then was poured into a dialysis bag (MW cutoff: 12000) and incubated in 100 ml of PBS solution in an orbital shaker at constant shaking of 50 rpm at 37 ℃ and pH 7.4. After predefined time intervals (0.5, 1, 2, 3, 6, 12, 24, 48 and 72 hours), 3 ml from aliquots of this suspension were taken and an equal volume of fresh PBS was compensated. The concentration of MTX was measured by spectrophotometry at λmax = 305 nm using a standard curve related to the absorption and MTX concentration, then the cumulative amount of released MTX was calculated. Similar to this condition, release from the optimized MTX loaded PLGA NPs was also investigated in slightly acidic milieu with pH 6.4. Control experiments using a free drug solution were performed to distinguish the difference between the release behavior of free drug and drug loaded NPs.
Human glioblastoma U87MG cells (obtained from Pasteur Institute of Iran, Tehran, Iran) was cultured in DMEM which is supplemented using 10% FBS and 100 µg/ml from penicillin-streptomycin at 37 ℃ in a humidified incubator with 5% CO2. Cells were maintained in an exponential growth phase by periodic dilutions with fresh medium. 1 × 104 viable cells were seeded in 100 ml of growth medium in 96-well plates and incubated 24 h to allow cell attachment. In order to determine the cytotoxicity effect of MTX loaded PLGA on human glioblastoma U87MG cells, this cells were incubated by the MTX (as drug control), blank PLGA NPs and MTX loaded PLGA NPs at different concentrations for 24, 48, 72 and 168 h. The effect of the different treatments on cell cytotoxicity was evaluated by MTT cytotoxicity assay. In brief, at the end of the incubation time with the different formulations, 100 μl from a MTT solution (5 mg/ml in PBS) was added to each well, and the culture medium containing MTT solution was removed after 3-4 h. 100 μl from Dimethyl Sulfoxide (DMSO) were then added to dissolve the formazan crystals. The UV absorbance of the solubilized formazan crystals was measured via a microplate reader (elx808 absorbance microplate reader) at 570 nm. The absorbance of the untreated cells was set at 100%. Cell viability and cytotoxicity percentage were calculated by the following equations:
Cell viability (%) = (Abs test cells/Abs control cells) × 100
Cytotoxicity (%) = 1-(Abs test cells/Abs control cells) × 100
Where Abs test cells is the optical density of the cells incubated with the different formulations and Abs control cells is the optical density of the cells incubated with the phosphate buffer (pH 7.4) as positive control (non-treated). IC50 was calculated by the curve fitting of the cell viability data using Prism 5.0 (Graph pad, USA) software.
All of the measurements were performed in triplicate. Data are presented as the mean ± standard deviation. A one-way Analysis of Variance (ANOVA) (SPSS 13.0, Chicago, USA) test was performed to determine the effect of various parameters on size distribution and EE. P < 0.05 was considered to be statistically significant.
A double-emulsion method was selected for preparation of the NPs due to the hydrophilic nature of MTX. Various parameters can affect the physicochemical properties of NPs including polymer concentration, PVA concentration in the external phase and drug concentration which play an effective role on the particle size.
PVA is a macromolecular emulsifier to prevent the aggregation and stabilize NPs of the emulsion [28]. Many of the hydroxyl groups of PVA were hydrated at the surface to stabilize NPs, and hydrocarbon chains of PVA were adsorbed on the surface of NPs [29]. The different concentrations of PVA were used to investigate its effects on the mean diameter of the NPs. The results indicated that particle size enhanced with increasing surfactant concentration and this increase was significant (p < 0.05) (Table 1).
Table 1: The effect of concentration of stabilizer on mean diameter of particles; n = 3. View Table 1
High concentration of PVA in the external phase may cause the inappropriate orientation of the chains on the surface of particles. Moreover, the interaction in aqueous environment enhanced as viscosity increased, thereby is not possible the fast binding to the surface to form small particles. PVA with 1% concentration was chosen for further studies because it prepares NPs with low and appropriate mean diameter.
To understand the influence of PLGA concentration on particle size, PLGA concentration in the internal phase increased from 1% to 4% w/v as seen in Table 2. Increasing the polymer concentration led to the larger particle size which was significant (P < 0.05). It can be explained that the increase in size is probably due to changes in viscosity and surface tension of the oil phase as well as increase in content of the solid mass of oil droplets which is similar to several studies [30-32].
Table 2: Influence of the PLGA concentration on mean diameter of particles; n = 3. View Table 2
As seen in Table 3, the particle size enhanced with increasing concentration of MTX and the increase was not significant (P > 0.05). The particle size changed from 184 nm to 286 nm as the amount of MTX increased from 5 to 20 mg. Using more amount of the drug can increase accumulation of the drug in the oil droplets, resulting in increase of the diameter. On the other hand, the problem of drug diffusion in the external aqueous phase is facilitated (which is not suitable for drug release).
Table 3: Physicochemical and Encapsulation properties of MTX Loaded PLGA nanoparticles, according to increase of drug value; n = 3. View Table 3
With optimizing synthesis process, MTX loaded PLGA NPs exhibited the mean size of 170 nm with range of 150 - 200 nm and narrow size distribution (PDI = 0.205) (Figure 1). The zeta potential of all NPs was low (Table 3). Moreover, SEM images (Figure 2) revealed that NPs shape was approximately spherical. These particles with diameter less than 200 nm for drug delivery purposes can have high potential for permeability to brain due to more diffusion in extracellular space of brain [33]. NPs with diameter less than 20 nm may be removed by kidney [34] whereas NPs with diameter more than 200 nm may be cleared using Reticuloendothelial Systems (RES) [35. Therefore, it seems that NPs with the diameter between 20 and 200 nm may be considered suitable.
 Figure 1: Particle size distributions of MTX Loaded PLGA NPs A) 153 nm; B) 161 nm; C) 196 nm. View Figure 1
Figure 1: Particle size distributions of MTX Loaded PLGA NPs A) 153 nm; B) 161 nm; C) 196 nm. View Figure 1
 Figure 2: SEM photograph of MTX loaded PLGA NPs.
View Figure 2
Figure 2: SEM photograph of MTX loaded PLGA NPs.
View Figure 2
EE and DL values in MTX loaded PLGA NPs were found to be dependent on the MTX concentration. The EE% and DL% of MTX loaded PLGA NPs enhanced by increasing the amount of MTX; however, the increase was not significant (P < 0.05) (Table 3). Therefore, increasing MTX from 5 to 20 mg in organic phase led to an increase in EE% from 62.69 to 71.21% and DL% from 5.72 to 8.22%. As the NPs prepared with 5 mg MTX were desirable in EE and DL, this concentration was used to obtain small NPs with appropriate EE and DL values. Moreover, by increasing the concentration of MTX, the amount of EE and DL did not change considerably whereas the size of NPs increased remarkably (Table 3). Therefore, it is expected that low concentrations of MTX may be more appropriate to obtain smaller particle size with the optimum amount of entrapped drug.
The FTIR spectra of PLGA, MTX and MTX loaded PLGA indicated that there was no change in the position of absorption peaks. Free MTX (Figure 3a) exhibit the absorption peaks at 3390, 2952 and 1700 cm-1 which are related to the presence of -OH, C-H and C = O groups. Main peaks of PLGA in 3433, 1767 and 1429 cm-1 (Figure 3b) can be attributed to -OH, C = O and C-O groups. The spectral analysis for MTX loaded PLGA NPs (Figure 3c) indicates that the functional groups in the NPs surface almost have the chemical characteristics similar to the PLGA and MTX. The FTIR spectra demonstrated that there was no chemical interaction between functional groups of MTX and polymer.
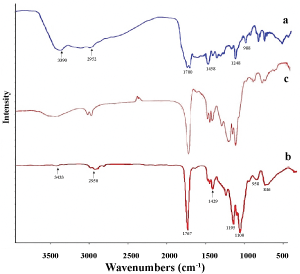 Figure 3: FTIR spectrums of a) MTX; b) PLGA; c) MTX loaded PLGA NPs. View Figure 3
Figure 3: FTIR spectrums of a) MTX; b) PLGA; c) MTX loaded PLGA NPs. View Figure 3
Significance of DSC provides information about physical properties of the samples including crystalline or amorphous nature. Figure 4 shows the DSC thermo grams of PLGA, MTX, physical mixture of MTX and PLGA, and MTX loaded PLGA NPs. The amorphous nature of PLGA indicated an endothermic peak at 45 ℃ corresponding to the polymer transition temperature (Tg). The pure MTX exhibited a single sharp endothermic melting peak at 120 ℃ and a wide exothermic peak at 240-260 ℃. The peaks of physical mixture of MTX and PLGA are similar to the peaks of pure MTX and PLGA. For MTX loaded PLGA NPs, the endothermic peak (45 ℃) of PLGA was observed whereas the peaks of MTX were not visible, indicating MTX entrapped into the NPs in an amorphous or disordered-crystalline phase.
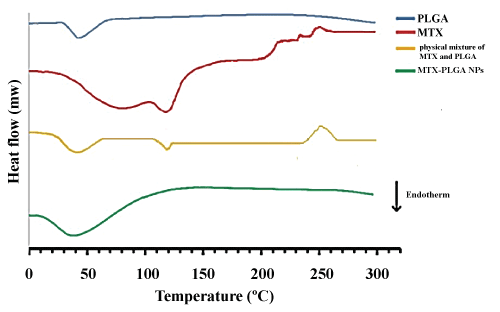 Figure 4: DSC thermogram of MTX, PLGA, physical mixture of MTX and PLGA, and MTX loaded PLGA NPs. View Figure 4
Figure 4: DSC thermogram of MTX, PLGA, physical mixture of MTX and PLGA, and MTX loaded PLGA NPs. View Figure 4
The in vitro drug release profiles of formulations are shown in Figure 5. In the control samples, free MTX exhibit that all drugs are released during 3 hours. MTX release profile from PLGA NPs at pH = 7.4 indicated that during the first 12 hours, about 30% of MTX was released from the NPs, which increased up to 50% in the first 24 hours followed by a sustained release up to 72 hours. In the first 24 hours, it seems that the drug adsorbed on or located on or close to the surface of the NPs was separated from the NPs into the aqueous phase. In the next hours, the release rate was slower due to medium penetration into the matrix and the whole mass of polymer and consequently continuous hydrolysis and degradation of polymers. The release of the drug continues until full dissolution of the polymer. This finding indicated a controlled behavior for release of MTX from NPs and a biphasic kinetic model for drug release. MTX release profiles from MTX loaded PLGA NPs at PH = 6.4 also indicated that the drug release rate increased by decreasing pH, and high value of the drug release occurred in the early hours. The reason can be attributed to acidic medium which affects the hydrolysis and degradation of polymer, resulting in the acceleration of the degradation of the polymer matrix which is in agreement with finding of Zolnik, et al. [36]. Therefore, MTX release from NPs in the medium of cancer cells and extracellular space cancer cells may have a pattern as these mediums have less pH level in comparison with buffered blood medium. Likewise, the in vitro drug release profiles of NPs revealed that MTX release rate can be controlled by the preparation of PLGA NPs formulation for an extended period of time.
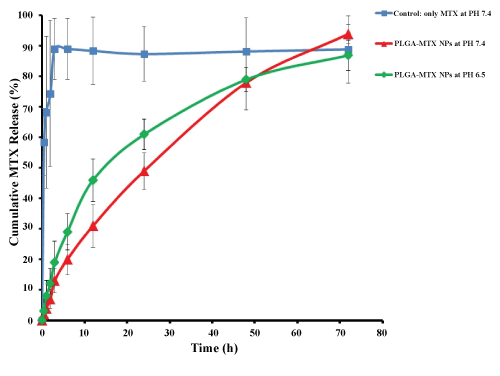 Figure 5: in vitro drug release profile of encapsulated MTX from PLGA NPs at 37 ℃. View Figure 5
Figure 5: in vitro drug release profile of encapsulated MTX from PLGA NPs at 37 ℃. View Figure 5
The cytotoxicity effect of both MTX loaded PLGA NPs and free MTX was concentration and time-dependent. Cell viability exceeded 95% in blank PLGA NPs for different incubation times, which indicated no cytotoxicity to the growth of U87MG cells. PLGA NPs exposed to U87MG cells showed very little cytotoxic effect even in high concentrations. The cytotoxicity effect of free MTX showed an increase in cytotoxic effect by increasing drug concentration whereas a decrease in the cytotoxicity was observed for all concentrations as incubation time increased. In 24 h incubation period (Figure 6A) cytotoxic effect of different concentration of free MTX was roughly similar and with increasing incubation time to 48 h, cytotoxicity enhanced and reached to maximum value of 35.2% (Figure 6B) whereas its cytotoxic effect in incubation time of 72 h specially in high concentrations decreased (Figure 6C). As seen in Figure 6D, in incubation time of 168 h, none of free drug concentration showed positive cytotoxic effects on U87MG cells which indicate ineffectiveness or lack of access of drug to cells. The results indicated that MTX loaded PLGA NPs had higher cytotoxic effect in all concentrations in comparison with free MTX.
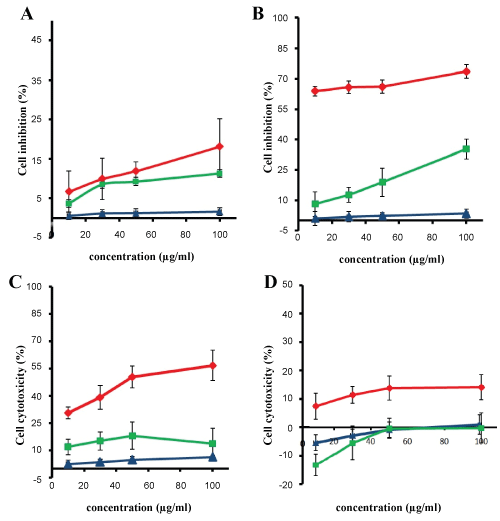 Figure 6: Inhibitory rate of U87MG cells treated with MTX loaded PLGA NPs (red), blank PLGA NPs (blue) and free MTX (green) (data are shown as the mean ± SD, n = 3). Incubation for A) 24 h; B) 48 h; C) 72 h; D) 168 h. View Figure 6
Figure 6: Inhibitory rate of U87MG cells treated with MTX loaded PLGA NPs (red), blank PLGA NPs (blue) and free MTX (green) (data are shown as the mean ± SD, n = 3). Incubation for A) 24 h; B) 48 h; C) 72 h; D) 168 h. View Figure 6
The different concentrations caused the significant effect on cell death and there is no significant difference between cytotoxic effects of high and low concentrations in same incubation time (Figure 7B). After 48 h of incubation, the NPs had highest value cytotoxicity (73.5%) which was nearly twice in comparison with time and similar concentrations of free drug cytotoxicity. For 48 h incubation times, no significant differences in cytotoxicity were observed among the different concentrations tested (Figure 7A). After 72 h, the cytotoxic effect of MTX loaded PLGA NPs reached to 56% and after 168 h, the cytotoxic effect of NPs reduced to approximately 15% (Figure 7B). This increase in cytotoxicity effects of incubation time of 48 and 72 h may be due to the accumulation of the significant amount of NPs in U87MG cells, leads to release of the sufficient values of MTX from structure of the NPs. For longer incubation times (168 h), free MTX lost its therapeutic effects whereas the NPs maintained the amount of the drug effects.
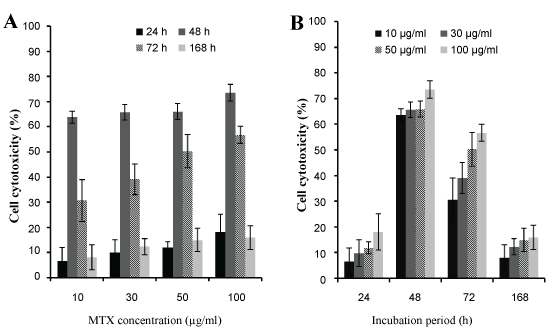 Figure 7: The cytotoxicity effect of A) Different concentrations of MTX loaded PLGA NPs; B) Different incubation time of MTX loaded PLGA NPs. View Figure 7
Figure 7: The cytotoxicity effect of A) Different concentrations of MTX loaded PLGA NPs; B) Different incubation time of MTX loaded PLGA NPs. View Figure 7
IC50 of free MTX with different incubation times is shown in Table 4. These values indicate that the IC50 of MTX in the MTX-PLGA NPs in comparison with the free MTX on glioblastoma U87MG cells significantly reduced and effect of nano-drug formulations was much more than pure drug. All these results could be attributed to the drug delivery carrier (PLGA), results in better transfer of drug and consequently improvement of the performance of the drug.
Table 4: IC50 values of free MTX and MTX Loaded PLGA NPs in different concentrations after incubation time of 24 h, 48 h, 72 h and 168 h with U87MG cells. View Table 4
The increased antitumor activity of MTX loaded PLGA NPs is due to the protection of the drug using PLGA from hydrolysis and decomposition as well as maintenance of the drug activity in a sustained release manner. Several pathways can be justified the increased antitumor activity of MTX incorporated into PLGA NPs:
1) Because of the enhanced endocytic activity of tumor cells, more NPs can be internalized into the cell which is consequently provided the greater concentration of drug [37]; 2) Increase in a concentration gradient near the cell surface may create a positive flow from drug into the cell [38]; 3) As MTX has similar structure to folate, the molecules of MTX on the surface of NPs may play a role in targeting NPs, leads to the increase in the transmembrane transport and cytotoxic effect of MTX loaded PLGA NPs [39]; 4) As glioblastoma cells have both cell resistant to chemotherapy and drug resistance transporter on their surface, the NPs have this ability to cross the cell membrane due to their small size and prevent drug resistance with sustained release drug inside the cell [40].
This study reports the preparation of MTX loaded PLGA NPs via double-emulsion solvent evaporation method for the delivery of the anticancer agent to human glioblastoma cells. The influence of different experimental parameters including polymer concentration, PVA concentration in the external phase and drug concentration on the particle size demonstrated that particle size enhanced with increasing PLGA and PVA and MTX concentration. Based on the optimized preparation method, the size of NPs was lower than 200 nm and the in vitro release profile of MTX from NPs indicated a sustained release behavior. The cytotoxic effect of MTX loaded PLGA NPs was more than free MTX. Moreover, MTX loaded PLGA NPs reduced the IC50 value of MTX loaded PLGA NPs for U87MG cells in comparison with free MTX.
This work was supported by Tehran University of Medical Sciences, Grant NO. 91-04-87-20209.