Iron is one of the elements that participate numerous reaction on the body and the structure of hemoglobin to the purpose of carrying oxygen to the tissues. Thus, iron deficiency causes different problems in the body. Iron-refractory iron deficiency anemia (IRIDA) is a genetic disorder that has some signs of iron deficiency anemia (IDA) but refractory to oral iron and partially refractory to intravenous iron. The mutation in TMPRSS6 gene causes matriptase-2 protein deficiency that negatively regulates hepcidin molecule. Thus, increasing hepcidin connects its receptor ferroportin and decreases iron absorption. Consequently, IRIDA appears.
Here we present a 7.5-years-old girl who was followed for three years with the clinical diagnosis of iron-refractory iron deficiency anemia, and mutations on TMPRSS6 gene.
After excluding the other iron deficiency causes, IRIDA and the mutations on TMPRSS6 gene should be kept in mind in patients with low transferrin saturation, normal levels of the ferritin, high levels of the hepcidin molecules and family history about iron deficiency anemia.
IRIDA, TMPRSS6 gene, Turkish population
IRIDA: Iron-Refractory Iron Deficiency Anemia; IDA: Iron Deficiency Anemia
Iron deficiency anemia (IDA) is a frequent entity that generates 50% of the anemia in the world [1]. Even if the etiologies of the IDA are often due to nutritional deficiency, bleeding or gastrointestinal malabsorption; genetically inherited iron deficiency anemia should also be kept in mind if there is an unknown cause of iron deficiency anemia.
Iron-refractory iron deficiency anemia (IRIDA) is a hypochromic microcytic anemia, an autosomal recessive disorder and shows iron deficiency anemia that is refractory to oral iron therapy but partially refractory to parenteral iron therapy. This hypochromic microcytic pattern connected with the low transferrin saturation, normal or high ferritin and abnormally high hepcidin levels [1]. The underlying reason for the disease is the mutations of the TMPRSS6 gene. Normally TMPRSS6 gene encodes a transmembrane serine protease matriptase-2 that has negative effect on the production of hepcidin iron regulatory protein. Hepcidin is the equalizer of the ferroportin, the main iron supplier. In TMPRSS6 gene mutation, matriptase-2 protein cannot be produced. Thus, hepcidin levels increase and inhibit ferroportin. Even though the iron storage exists, the iron that cannot enter to the systemic circulation causes iron deficiency anemia and this anemia is refractory to oral iron therapy.
Here we present a patient diagnosed as iron deficiency anemia refractory to iron and confirmed with the TMPRSS6 gene analysis.
7.5-years-old girl admitted with the complaints of paleness, weakness and growth retardation. Although no convenient history was taken from patient's mother, it is learned that she was diagnosed with alpha thalassemia in her early ages and was given intravenous iron therapy when she was 5-years-old because she was not responding to oral iron therapy.
In her physical examination, weight 22 kg (25-50 percentage), height 119.5 cm (25 percentage), slightly protruding facial bones, mildly thalassemic and pale facial appearance were revealed. There was no organomegaly. The other systemic findings were normal.
Because of the complaint of the growth retardation, the patient's growth curve analyzed and it showed that weight and height development were within normal ranges showing the same pattern through years.
According to the patient's first laboratory assessment the values were as follows: Erythrocyte count 5.21 × 106/µL (3.9-6.2 × 106/µL), Hemoglobin 9.7 g/dL (11.5-14.5 g/dL), Hematocrit 32.4% (35-45%), Mean Corpuscular Volume 62.2 fL (77-87 fL), Mean Corpuscular Hemoglobin 18.6 pg (25-29 pg), Mean Corpuscular Hemoglobin Concentration 29.9 g/dL (32-36 g/dL), RDW 17.7% (11.6-14.8%). There was hypochromia, anisocytosis, microcytosis and poikilocytosis in peripheral blood smear examination.
Iron concentration and transferrin saturation were low, whereas iron binding capacity and ferritin were found in normal levels. In further examination 25-hydroxy vitamin D level was found low but the other laboratory analysis were found normal (Table 1).
Table 1: Laboratory findings. View Table 1
Hemoglobin electrophoresis was within normal levels with HbF 0.3% (0-2%), HbA2 2.4% (0-4%), HbA 97.3% (94-99%). There were no alpha thalassemia mutations in alpha thalassemia gene research. In the point of celiac disease, endomysial antibody, IgG, and tissue transglutaminase IgA were also found negative.
Iron absorption test revealed a flat curve showing iron absorption defect (Figure 1). As there is a close relation with iron and zinc at the gastrointestinal level, as reported previously, we performed oral zinc tolerance test [2,3] (Figure 2). Patient's oral zinc tolerance test was found to be normal and serum zinc levels showed a higher peak compared to controls. Due to these findings pre-diagnosis was thalassemia trait, with iron deficiency anemia.
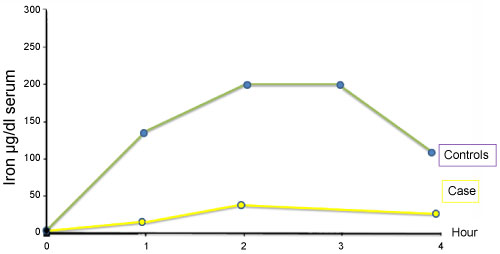 Figure 1: Iron absorption test.
View Figure 1
Figure 1: Iron absorption test.
View Figure 1
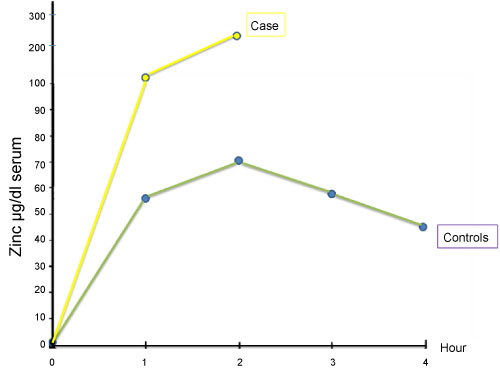 Figure 2: Zinc absorption test.
View Figure 2
Figure 2: Zinc absorption test.
View Figure 2
In abdominal ultrasonography; there was millimetric crystalloid in left kidney, minimal irregularity on the bladder wall, mobile echogenic materials in lumen and debris materials that are accumulated on bladder base. Apart from that; liver, gallbladder, spleen, pancreas, adrenal glands, abdominal aorta and vena cava inferior were found normal. There was no lymphadenopathy in pathological size, a mass and free fluid in Douglas. It was stated that uterus and ovary are compatible with age.
The patient had urinary tract infections two times in her early childhood but did not repeated later on. Her urine examination was normal and culture was negative. Patient's grandfather has urolithiasis and her aunt has gravel, therefore the patient is followed in this respect. Plenty of liquid and nutrition advice were given to the patient and 3 months later USG repetition is planned.
After all of these results were evaluated and the other provisional diagnoses were excluded, it is thought that it can be a genetic disorder of the iron metabolism. Thereupon, as a result of the examinations done, the patient was diagnosed with the IRIDA with the mutations in TMPRSS6 gene. TMPRSS6 gene in compound heterozygosity p.G603R (c.1807G > C) and c.1904_1905dup (p.K636AfsX17) were detected in DNA sequence analysis in the IRIDA-mediated TMPRSS6 gene.
Although it is told that the IRIDA prevalence is 1/100.000, it should be more frequent because of the lack of the diagnosis. Up to today, 58 different mutations are detected in the TMPRSS6 gene in 51 families and 74 patients who have different etiologies [1]. Among these (p.Lys636fs; p.Lys636fs), (p.Gln571Glnfs; p.Gln571Glnfs), (p.Arg599X; p.Arg599X), (IVS10 + 1 G > A; p.Cys510 > Arg) mutations were determined in Turkish population [4-6].
G603R mutation in one allel and K636A mutation in the other was detected in our case. Although these mutations were reported in the other populations, these two were reported for the first time in Turkish population [5,7].
IRIDA is an iron absorption deficiency which is an autosomal recessive disorder caused by mutations on TMPRSS6 gene. Although the cases reported are less than 100, it is thought that IRIDA is the most frequent cause of the “atypical” hypochromic microcytic anemia [4].
After excluding the other iron deficiency causes, IRIDA should be kept in mind in patients with low transferrin saturation, normal levels of the ferritin, high levels of the hepcidin molecules and family history about iron deficiency anemia.
Although the standard therapy, iron deficiency anemia occurs without any obvious loss of iron in IRIDA and it repeats after the parenteral iron therapy and it is also partially refractor to this therapy.
In the case of the existence of these signs, IRIDA and the mutations on TMPRSS6 gene should come to mind. Several diseases can be figured out with the process of starting from the cure and going to the diagnosis. Holistic view is worthwhile.
Each author have participated sufficiently in the work to take public responsibility for the content and also the authors had no conflict of interest to declare in relation to this manuscript. As it was a retrospective study, ethics approval was not taken.
The authors can confirm that all relevant data are included in the manuscript.
The authors declare that there are not any competing interests in relation to this work.
No funding was received.
Yasemin ARDICOGLU AKISIN (Drafting, reviewing), Gülnaz KURT (Drafting), Huseyin ONAY (Sequence analysis), Ferda OZKINAY (Sequence analysis), Nejat AKAR (Clinical Diagnosis, reviewing).