

International Journal of Brain Disorders and Treatment
Pathological Changes of Astrocytes under Seizure
Shanshan Lu1, Fushun Wang1,2,3* and Jason H Huang3*
1Department of Psychology, Nanjing University of Chinese Medicine, China
2Department of Neurosurgery, University of Rochester, USA
3Department of Neurosurgery, Baylor Scott & White Health, USA
*Corresponding author:
Fushun Wang and Jason H. Huang, Department of Neurosurgery, Baylor Scott & White Health, Temple, Texas 76508, USA, E-mail: fushun_wang@urmc.rochester.edu, jhuang@sw.org
Int J Brain Disord Treat, IJBDT-2-008, (Volume 2, Issue 1), Review Article; ISSN: 2469-5866
Received: December 14, 2015 | Accepted: January 07, 2016 | Published: January 09, 2016
Citation: Lu S, Wang F, Huang JH (2016) Pathological Changes of Astrocytes under Seizure. Int J Brain Disord Treat 2:008. 10.23937/2469-5866/1510008
Copyright: © 2016 Lu S, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Multiple lines of studies support the view that defective functions of astrocytes contribute to neuronal hyper-excitability in the epileptic brain. Autopsy and surgical resection specimens find that post-traumatic seizures and chronic temporal lobe epilepsy may originate from glial scars. Astrogliosis, a component of glial scar, which involves structural and metabolic changes in astrocytes, is often a prominent feature of temporal epilepsy and most animal models of recurrent seizures. Although glial scar formation has been recognized for over 120 years, fundamental aspects of the cellular mechanisms are poorly understood. We here provide an overview of the experimental findings about astrogliosis in seizures, which involves the changes in astrocytic structure and loss of domain. These structure changes are paralleled by functional changes, including expression levels of glutamate transporter and glutamine synthetase. Reactive astrocytes have also been shown to down-regulate the activities of K+ channels, leading to impairment of clearance of K+. The functional remodeling may contribute to increased neuronal excitability and generation of seizures.
Keywords
Astrocyte, Astrogliosis, Seizure, Potassium, Glial scar
Introduction
Epilepsy is a common chronic neurologic disorder affecting at least 50 million people [1,2], or more than 1% of the population worldwide [3-5]. Epileptic seizures are uncontrolled sudden attacks of a non-convulsive or convulsive nature associated with intensive neuronal firing. Spatially restricted foci in seizure brain can often be identified for epilepsies acquired after tumors, head trauma, or other severe focal brain, insults brain [6]. The cellular or molecular mechanisms underlying epileptic form activity have not been fully understood, but the contributions of astrocytic cells have been suggested by many studies.
Astrocytes are the most abundant glial cell type in the brain [7], which are characterized by leaflet-like processes and very irregularly shaped cell bodies, and cover almost all excitatory synapses in the brain [7,8]. Historically, glial cells were only thought to provide neurons with only metabolic and physical supports, to control the ionic homeostasis, and control the neuronal excitability through K+ buffering, and serve as the primary energy source for neurons [9,10]. Current research has expanded their function to important homeostatic and neuronal modulatory functions [11]. Astrocytes are involved in regulating ion homeostasis, maintenance of the blood brain barrier (BBB) function, metabolism of amino acid neurotransmitter, as well as nutrient and energy support for neurons [12]. Our recent reports demonstrated that astrocytes could actively regulate these processes with intracellular Ca2+ wave regulated signaling pathways [13,14]. The present review summarized the roles of reactive astrocytes in the development and progression of epileptic seizures, and discussed the relevance of calcium signaling in astrocytes to therapeutic management of the disease.
Astrogliosis after Brain Injury
Almost all traumatic brain injury (TBI), including prolonged seizures, results in reactive gliosis (astrogliosis), which are characterized by severe biochemical and morphological changes of pre-existing astrocytes and also new astrocyte generation. These astrocytes constitute a dense scar tissue that has been suggested to separate the injured tissue from its surroundings and demarcate the lesion area. Experimental ablation of astrocytes after TBI results in larger lesions, worse demyelination, local tissue disruption, and exacerbated neuron death. Activated astrocytes is beneficial in that they produce a barrier against the spread of neurotoxicity and prevent excitatory amino acid induced cell death [15]. The glial scar consists predominately of microglia, reactive astrocytes, and extracellular matrix molecules, such as chondroitin sulfate proteoglycans. The reactive astrocytes provide metabolic and tropic support to prevent secondary degeneration. In addition, astrocytes provide tropic support at the injury site which promotes the survival of nearby neurons. Though astroglial scars are essential for wound repair, they also interfere with axonal regrowth [16]. Their effects of inhibiting axon regrowth have been studied and described in considerable mechanistic and descriptive detail. For instance, it was found that the astrocytes are oriented perpendicular to lesions during scar border formation, and they quantifiably reorient to become more or less parallel to the lesion as scar formation progresses. And it is found that the initial perpendicular orientation of the elongated astrocytes appear remarkably similar to that of palisading astrocytes perpendicular to cortical lesions after TBI and implicated in post TBI epileptic foci [17]. In addition, the mechanisms of glial scar inducing seizure are also possibly due to astrogliosis, with its ability of K+ buffering and neurotransmitter clear up decreased.
Cytokines Released by Reactive Astrocytes
Although glial scar formation has been recognized for over 120 years, fundamental aspects of the cellular mechanisms, molecular regulation, and adaptive function underlying astrogliosis remain poorly understood [16]. Lesion-derived diffusible inflammatory factors such as cytokines, growth factors might be involved, for signaling mechanisms that determine astrogliosis have for some time been associated with penetrating CNS lesions that disrupt the BBB, instead of CNS insults where BBB function is preserved; and astrogliosis varies according to the distance from lesions [16]. Dysfunction of the BBB is a hallmark of brain insults and usually surrounds the core lesion. Focal epilepsy typically arises either within or adjacent to a cortical lesion [18].
Astrocytes can produce many pro- and anti-inflammatory molecules and have dynamic functions. The production of TGF-β and TNF-α by astrocytes can lead to detrimental effects depending on the receptors activated and expression timing [19]. TGF-βs play a critical role in the intercellular communication, and are involved in embryogenesis, cell growth, would healing, morphogenesis, and apoptosis in a wide variety of cells [18]. TGF-β1 expression is up-regulated in the brains suffering from Alzheimer's disease, stroke, multiple sclerosis, tumor or traumas. TGF-βs have also been found to be elevated in the cerebro-spinal fluid (CSF) of patients with brain injury [20]. The preferentially uptake of serum albumin by glial cells within hours following brain injury in prior to the seizure development [21], raised the hypothesis that glial functions and dysfunctions play a critical role in the epileptic generation [18]. Aronica et al further reported that the up-regulated expression of TGF-β in astrocytes from the hippocampus of SE-experienced rats [22].
Multiple inflammatory factors induced by pathologically TBI induced changes have been described, for example, growth factors such as TNF-α, TGF-β can induce changes in neuronal calcium homeostasis. The release of TNF-α, TGF-β could be released by astrocytes in responses to excitotoxic injection. The epileptic brain can also be induced by astrocytic dys-regulation of the vascular system. The up-regulation or induction of IL-1β, complement components, and many other inflammatory factors within perivascular astrocytes, may have dramatic effects on the vascular system leading to disruption of the BBB [23]. Recent studies from several laboratories confirmed the role of vascular pathology in the epileptic development and demonstrated a critical role for astrocytes in epilepsy. Mechanical stimulation under pathological conditions could provide the injured CNS with impaired domain, mass effects, swelling, or tissue hypertrophy [24].
Reactive astrocytes are characterized by up-regulation of glial fibrillary acidic protein (GFAP) expression and significant hypertrophy of cell bodies and processes. Interestingly, the severity of astrocygliosis is often a predictor of post TBI seizure, and intra-operative recordings have found that post TBI seizures and temporal lobe epilepsy are often initiated in, or immediately adjacent to glial scar tissue (McKhann 2000). Our previous studies found that a marked increase in overlap of processes between adjacent astrocytes after brain injury (Figure 1). There are two distinct types of reactive astrocytes in animals with focal epilepsy: hypertropic astrocytes, which located just beyond the annular sector occupied by palisading astrocytes; and palisading astrocytes, which immediately surround the injury site, whose processes form a halo around the lesion with striking radial orientation. The hypertropic astrocytes plays an important role in pathogenesis of seizures by releasing the cytokines, and also exert a continued deleterious effect on tissue [25]. Palisading astrocytes exhibited a complete loss of individual domains and appeared to form a physical barrier separating the lesion from the surrounding cortical tissue. Astrocytes are possibly organized in non-overlapping domains, and the loss of domain due to astrocytic structure changes are paralleled by functional changes, such as expression levels of glutamine synthetase and glutamate transporter.
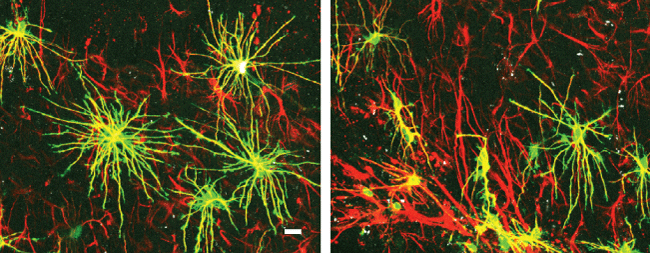
.
Figure 1: Loss of astrocytic domain organization after brain injury.
Left: astrocyte in normal conditions; Right: new astrocytes and normal astrocytes at the glial scar mixed together, demonstrating loss of the domain organization, modified from Oberheim et al [17]. Green, GFAP; red, DiD; scale bar = 20 μm.
View Figure 1
Astrocytic Modulation of Glutamate
The astrocytic function of housekeeping is to maintain a low concentration of extracellular glutamate and prevent excitotoxicity. Astrocytic uptake of glutamate terminates the effects of glutamate as a neurotransmitter [26,27]. And it is commonly accepted that GLT-1 transporters (excitatory amino acid transporter-2/EAAT2) in astrocytes play a large role in glutamate uptake. High affinity glutamate transporters, GLAST1 and GLT1 subtype, are enriched in astrocytic processes, and play the predominant role for glutamate clearance in the brain. Glutamate transporting is an electrogenic and energy demanding process, with one glutamate being co-transported with three Na+. And then glutamate is converted to glutamine via an ATP-consuming process, catalyzed by a glutamine synthetase, an astrocyte-specific enzyme, in the astrocytes. Glutamine is subsequently released to fuel the neurons and recycled into glutamate for glutamatergic neurotransmission. Glutamine synthetase (GS) plays a critical role in the glutamate metabolism [26], and glutamine synthetase has been shown to be up-regulated in reactive astrocytes. But it appears to be reduced in the epileptic hippocampus, indicating potential changes in the astrocytic transporters. Similiar change of glutamate transporters have similarly been shown to be reduced in the epileptic hippocampus [28], which might be due to Na+, K+-ATPase impairment affected by high extracellular K+.
It is now reported that peri-synaptic astrocyte may detect spillout of glutamate and other substances from active synapses under seizures and respond structurally by modifying and extending their processes. Astrocytes respond to synaptic neurotransmitters release with both Ca2+ waves and glio transmitter release that can further enhancing seizures, with important implications in the epileptic brain [29]. In addition, astrocytes can also modulate synaptic function and modulate inter-synaptic crosstalk via regulation of extracellular glutamate diffusion. In addition, glutamate can interact with mGLuR to induce astroglial cell swelling by modulating intracellular Ca2+, which can be detected simultaneously with changes in cell volume. Expressions of the EAAT2 transporters are significantly decreased in CA1 region in the sclerotic human hippocampus. In addition, sclerotic brain tissues removed during epilepsy surgery are characterized by down-regulation of glutamate synthetase in astrocytes [30]. The glutamate cycle is the major way to remove released glutamate from extracellular space. Following its synaptic release from the glutamatergic neuron terminals, glutamate is transported into astrocytes where it is converted into glutamine by glutamine synthase; glutamine is exported and taken up by neurons, where it is converted back to glutamate by mitochondrial glutaminase. Indeed, glutamate-glutamine cycling appears to be slowed in sclerotic hippocampus removed during epilepsy surgery [31]. With this process slowing down, intracellular glutamate might be increased and released into extracellular space, and astrocytic glutamate signaling might be enhanced. There is no question that glutamate can act as a key transmitter of bidirectional communication between astrocytes and neurons, and many studies have pointed out that astrocytes can release glutamate. Previous studies in our lab suggested the role of astrocytes in initiation, maintenance and spread of epileptiform activities by astrocytic glutamate release (Figure 2) [32]. Synchronized population spikes are key concomitants to seizure. Some studies have indicated multi-synaptic excitatory pathways can trigger synchronized burst activity in picrotoxin (GABAA receptor blocker)-induced seizure activity, while some other evidence pointed to the roles of both recurrent inhibition and gap-junction coupling. Studies in our lab suggested that an action-independent source of glutamate can trigger local depolarization events and synchronized bursting activity.
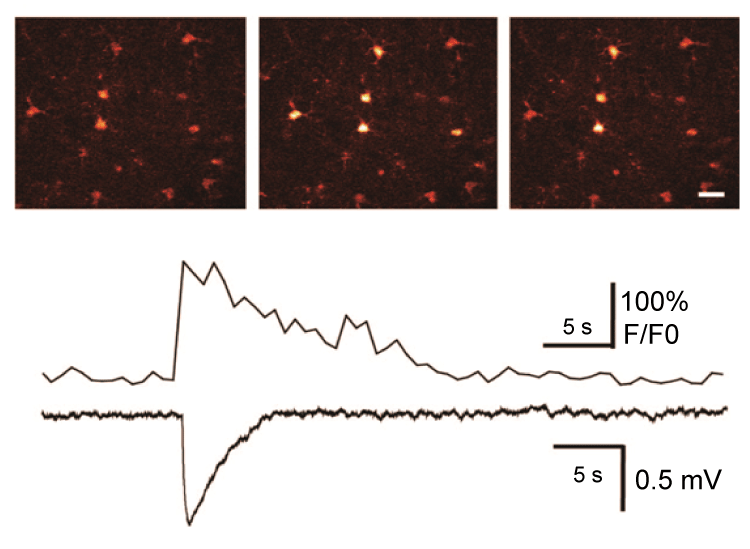
.
Figure 2: Photolysis of caged Ca2+ (NP-EGTA) in an astrocyte elicits a local depolarization shift in the presence of 1 μM TTX. Upper panel: Sequence of pseudocolor images of an astrocyte loaded with NPEGTA/AM and fluo-4/AM. Delivery of UV pulses targeting the astrocyte elevates cytosolic Ca2+ and triggers a spontaneous depolarization shift. Scale bar, 20 μm. Lower panel: traces of astrocytic Ca2+ concentration and field potential, and depolarization shift. Modified from Tian et al. [32].
View Figure 2
Increase in [K+]O can Induce Paroxysmal Oscillation
It is found that K+ buffering is impaired in sclerotic tissue, by comparison of hippocampal slices obtained from patients without or with or mesial temporal sclerosis [33]. Impaired astrocytic K+ buffering is expected to result in slower K+ clearance, lower seizure threshold and thereby contributing to seizure generation. The concentration of extracellular K+ is an important factor that regulates the neuronal excitability in the neural network [9,10]. Extracellular K+ accumulation is considered to be a favorable condition for the seizure onset, and impaired K+ uptake by astrocytes might be the initiation of epileptiform activity in the hippocampus. It is estimated that the passage of a single action potential can trigger 1 mM transient increase in extracellular K+( [K+]o ), above the ~3 mM resting level. Intensive neuronal stimulation causes nearly 5 mM an increase above the resting level. Peaks of 10-12 mM K+ can be reached during hyper-synchronous neuronal activities that characterized epileptic disorders [29,34]. Increased K+ due to intense neuronal firing discharges can depolarize neurons and facilitate the development of epileptiform discharges [35]. However, even though it is well established that the extracellular [K+]o concentration increases during seizure, but whether astrocytic buffering imparing is the primary seizure-eliciting factor remains unknown. Astrocytes play an essential role in maintenance of extracellular K+ at a level compatible with normal neuronal activity [11]. In vitro studies have shown that stimulation-induced K+ increases are paralleled by K+ accumulation in astrocytes, suggesting the roles of astrocytes in K+ regulation [36]. The level of K+ was determined by K+ mediated currents, glial buffering and K+ pump [14]. Our studies also suggested that agonist induced Ca2+ wave can facilitate the extracellular K+ uptake (Figure 3) [13,14,37], but when either K+ pump or glial buffering failed to act normally, the K+ increase leads to paroxysmal bursting. In addition, with K+ buffering ability decreases, many other functions might also be affected, such as intracellular Cl- will also be increased. With intracellular Cl- increase, the inhibition pathway was impaired [38].
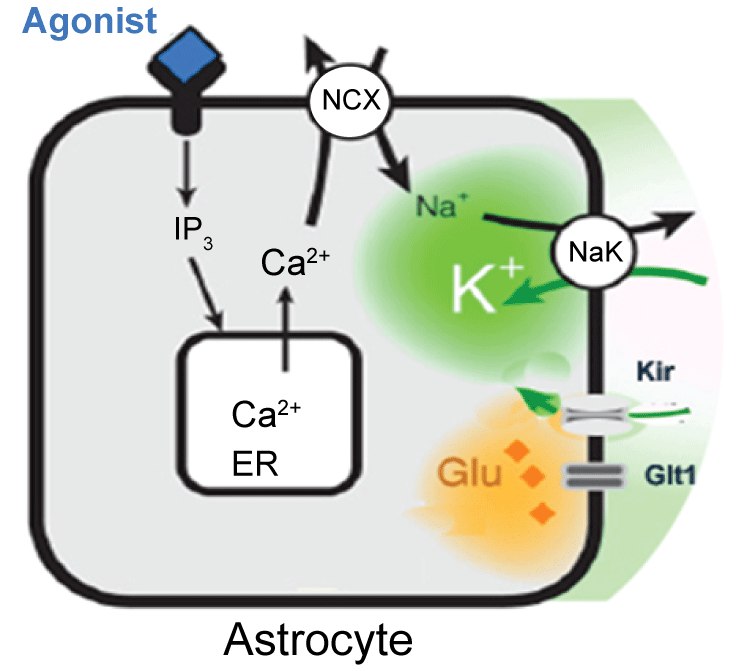
.
Figure 3: Agonist induced activation of astrocytic Gq-linked receptors triggers intracellular IP3 release and mobilization of Ca2+ from endoplasmic reticulum. In turn, the Na+, Ca2+ exchanger transports Ca2+ on the expense of Na+ influx. The increase in cytosolic Na+ activate Na+/K+-ATPase, resulting in a decrease in extracellular K+. Modified from Wang et al. [14].
View Figure 3
Conclusion
Epilepsy is a common chronic neurologic disorder affecting ~1% of the population with an estimated lifetime risk greater than 4%. In spite of optimal medical management, many patients with epilepsy remain medically refractory and suffer from debilitating seizures. Treatment options for these patients include new antiepileptic drugs, which target ion channels, such as Na+, Ca2+ channels or GABA receptors. These drugs are only effective in a small percentage of patients, not as effective as resective surgery which are effective in 60~80% of patients. So far there are few treatments targeting astrocytes, even though many studies pointed to astrocytic cells. Historically, astrocytes were thought to provide only physical and metabolic support for neurons, serving as the primary energy source for neurons and maintaining the water and ionic homeostasis and buffering K+. In addition, it is now clear that astrocyte can release of gliotransmitters in response to synaptic neurotransmitters induced Ca2+ waves and that in turn can further influence synaptic activity, with important implications in the epileptic brain [29]. Astrocytes play a critical role in glutamate uptake, mostly through the GlT-1 transporter, with assistance by additional uptake via the EAAT1 transporters. Glutamine synthetase plays a key role in the metabolism of uptaken glutamate. In addition, the major housekeeping functions of astrocytes include the maintenance of extracellular K+ homeostasis [37]. Some investigators reported changes in the pathological changes of glial cells in epileptic scar tissue, and these astrocytes failed to take K+ [39]. In conclusion, astrocytes are highly complex cells, and function to support the neuronal microenvironment and to modulate the excitability of neural networks under seizures [40]. Many properties of astrocytes also make them important targets for the developing field of treatment of epilepsy.
Conflicts of Interest
The authors declare that there are no competing financial interests.
Acknowledgements
This work was supported, in part, by NIH R01 NS067435 (J.H.H.), Scott & White Plummer Foundation Grant (J.H.H.), Jiangsu Specially Appointed Professorship Foundation (F.W.), Jiangsu Nature Foundation BK20151565 (F.W.), Jiangsu Traditional Chinese Medicine Foundation ZD201501 (F.W.), Jiangsu Six Talent Peak (2015-YY-006), and also the priority academic program development of Jiangsu Higher Education Institute PAPD (F.W.). We would like to thank Maiken Nedergaard for her input and assistance for this study.
References
-
Baldin E, Hauser WA, Buchhalter JR, Hesdorffer DC, Ottman R (2014) Yield of epileptiform electroencephalogram abnormalities in incident unprovoked seizures: A population-based study. Epilepsia 55: 1389-1398.
-
Witcher MR, Ellis TL (2012) Astroglial networks and implications for therapeutic neuromodulation of epilepsy. Front Comput Neurosci 6: 61.
-
Ngugi AK, Bottomley C, Kleinschmidt I, Sander JW, Newton CR (2010) Estimation of the burden of active and life-time epilepsy: a meta-analytic approach. Epilepsia 51: 883-890.
-
Thurman DJ, Beghi E, Begley CE, Berg AT, Buchhalter JR, et al. (2011) Standards for epidemiologic studies and surveillance of epilepsy. Epilepsia 52: 2-26.
-
Losi G, Cammarota M, Carmignoto G (2012) The role of astroglia in the epileptic brain. Front Pharmacol 3: 132.
-
Wetherington J, Serrano G, Dingledine R (2008) Astrocytes in the epileptic brain. Neuron 58: 168-178.
-
Oberheim NA, Takano T, Han X, He W, Lin JH, et al. (2009) Uniquely hominid features of adult human astrocytes. J Neurosci 29: 3276-3287.
-
Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, et al. (1994) Glutamate-mediated astrocyte-neuron signalling. Nature 369: 744-747.
-
Kofuji P, Newman EA (2004) Potassium buffering in the central nervous system. Neuroscience 129: 1045-1056.
-
Ma B, Buckalew R, Du Y, Kiyoshi CM, Alford CC, et al. (2015) Gap junction coupling confers isopotentiality on astrocyte syncytium. Glia.
-
Seifert G, Steinhäuser C (2013) Neuron-astrocyte signaling and epilepsy. Exp Neurol 244: 4-10.
-
Verkhratsky A, Nedergaard M, Hertz L (2015) Why are astrocytes important? Neurochem Res 40: 389-401.
-
Wang F, Xu Q, Wang W, Takano T, Nedergaard M (2012) Bergmann glia modulate cerebellar Purkinje cell bistability via Ca2+-dependent K+ uptake. Proc Natl Acad Sci U S A 109: 7911-7916.
-
Wang F, Smith AN, Xu Q, Goldman S, Peng W et al. (2013) Photolysis of caged Ca2+ but not receptor-mediated Ca2+ signaling triggers astrocytic glutamate release. J Neurosci 33: 17404-17412.
-
Rolls A, Shechter R, Schwartz M (2009) The bright side of the glial scar in CNS repair. Nat Rev Neurosci 10: 235-241.
-
Wanner IB, Anderson MA, Song B, Levine J, Fernandez A, et al. (2013) Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J Neurosci 33: 12870-12886.
-
Oberheim NA, Tian GF, Han X, Peng W, Takano T, et al. (2008) Loss of astrocytic domain organization in the epileptic brain. J Neurosci 28: 3264-3276.
-
Heinemann U, Kaufer D, Friedman A (2012) Blood-brain barrier dysfunction, TGFß signaling, and astrocyte dysfunction in epilepsy. Glia 60: 1251-1257.
-
Wilde GJ, Pringle AK, Sundstrom LE, Mann DA, Iannotti F (2000) Attenuation and augmentation of ischaemia-related neuronal death by tumour necrosis factor-alpha in vitro. Eur J Neurosci 12: 3863-3870.
-
Phillips DJ, Nguyen P, Adamides AA, Bye N, Rosenfeld JV, et al. (2006) Activin a release into cerebrospinal fluid in a subset of patients with severe traumatic brain injury. J Neurotrauma 23: 1283-1294.
-
Ivens S, Kaufer D, Flores LP, Bechmann I, Zumsteg D, et al. (2007) TGF-beta receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain 130: 535-547.
-
Aronica E, van Vliet EA, Mayboroda OA, Troost D, da Silva FH, et al. (2000) Upregulation of metabotropic glutamate receptor subtype mGluR3 and mGluR5 in reactive astrocytes in a rat model of mesial temporal lobe epilepsy. Eur J Neurosci 12: 2333-2344.
-
Aronica E, Sisodiya SM, Gorter JA (2012) Cerebral expression of drug transporters in epilepsy. Adv Drug Deliv Rev 64: 919-929.
-
Ostrow LW, Sachs F (2005) Mechanosensation and endothelin in astrocytes--hypothetical roles in CNS pathophysiology. Brain Res Brain Res Rev 48: 488-508.
-
Morcos Y, Lee SM, Levin MC (2003) A role for hypertrophic astrocytes and astrocyte precursors in a case of rapidly progressive multiple sclerosis. Multiple sclerosis 9: 332-341.
-
Coulter DA, Eid T (2012) Astrocytic regulation of glutamate homeostasis in epilepsy. Glia 60: 1215-1226.
-
Giménez-Cassina A, Martínez-François JR, Fisher JK, Szlyk B, Polak K, et al. (2012) BAD-dependent regulation of fuel metabolism and K(ATP) channel activity confers resistance to epileptic seizures. Neuron 74: 719-730.
-
Proper EA, Hoogland G, Kappen SM, Jansen GH, Rensen MGA, et al. (2002) Distribution of glutamate transporters in the hippocampus of patients with pharmaco-resistant temporal lobe epilepsy. Brain 125: 32-43.
-
Carmignoto G, Haydon PG (2012) Astrocyte calcium signaling and epilepsy. Glia 60: 1227-1233.
-
Eid T, Thomas MJ, Spencer DD, Runden-Pran E, Lai JC, et al. (2004) Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet 363: 28-37.
-
Petroff OA, Errante LD, Rothman DL, Kim JH, Spencer DD (2002) Glutamate-glutamine cycling in the epileptic human hippocampus. Epilepsia 43: 703-710.
-
Tian GF, Azmi H, Takano T, Xu Q, Peng W, et al. (2005) An astrocytic basis of epilepsy. Nat Med 11: 973-981.
-
Heinemann U, Gabriel S, Jauch R, Schulze K, Kivi A, et al. (2000) Alterations of glial cell function in temporal lobe epilepsy. Epilepsia 41(s6): S185-S189.
-
Walz W (2000) Role of astrocytes in the clearance of excess extracellular potassium. Neurochem Int 36: 291-300.
-
Timofeev I, Bazhenov M, Sejnowski T, Steriade M (2002) Cortical hyperpolarization-activated depolarizing current takes part in the generation of focal paroxysmal activities. Proceedings of the National Academy of Sciences of the United States of America 99: 9533-9537.
-
Larsen BR, Assentoft M, Cotrina ML, Hua SZ, Nedergaard M, et al. (2014) Contributions of the Na+/K+-ATPase, NKCC1, and Kir4.1 to hippocampal K+ clearance and volume responses. Glia 62: 608-622.
-
Wang F, Smith NA, Xu Q, Fujita T, Baba A, et al. (2012) Astrocytes modulate neural network activity by Ca2+-dependent uptake of extracellular K+. Sci Signal 5: ra26.
-
Rangroo Thrane V, Thrane AS, Wang F, Cotrina ML, Smith NA, et al. (2013) Ammonia triggers neuronal disinhibition and seizures by impairing astrocyte potassium buffering. Nat Med 19: 1643-1648.
-
Crunelli V, Carmignoto G (2013) New vistas on astroglia in convulsive and non-convulsive epilepsy highlight novel astrocytic targets for treatment. J Physiol 591: 775-785.
-
Simard M, Nedergaard M (2004) The neurobiology of glia in the context of water and ion homeostasis. Neuroscience 129: 877-896.
