Frank-ter Haar syndrome is a genetic disease that is transmitted by autosomal recessive pattern with characteristic features such as megalocornea or glaucoma, a developmental delay and multiple congenital anomalies. It was first recognized as a separate entity by Frank, et al. [1] and subsequently confirmed by ter Haar, et al. [2]. The main characteristics are brachycephaly, wide fontanels, prominent forehead, hypertelorism, prominent eyes, megalocornea with or without glaucoma, full cheeks, small chin, bowing of the long bones, and flexion deformity of the fingers. Protruding, simple ears, and prominent coccyx bone can be also regarded as important diagnostic signs. Inheritance most likely is autosomal recessive. Several manifestations such as progressive "coarsening" of the face, hirsutism, gallstones, lingual papillomatosis, and cardiac valve anomalies all point to a possible metabolic basis of the disorder. Here we describe an Iranian 6-year-old boy with this syndrome which is confirmed by a homozygous pathogenic mutation in the SH3PXD2B gene.
ter Haar syndrome, Frank-ter haar syndrome, Hypertelorism, Mitral valve insufficiency, Kyphoscoliosis, Skeletal dysplasia, Autosomal recessive, Filamin A
Frank, et al. [1] described an 18-month-old Bedouin girl, born to consanguineous parents, with megalocorneae, multiple skeletal anomalies, and developmental delay, and suggested that this combination of anomalies was a hitherto unreported entity. ter Haar, et al. [2] reported on three Dutch patients from one family with craniofacial and skeletal characteristics resembling Melnick-Needles syndrome (MNS) but with a fatal course and autosomal recessive inheritance. In 1995, another affected boy, with a similar phenotype, was described from the same but extended family as studied [2,3]. These authors suggested that this was an entity distinct from MNS and proposed the eponym ter Haar syndrome. As the patient described by Frank, et al. [1] shows a remarkable resemblance to the patients described by ter Haar, et al. [2], it seems very probable that both patients have the same entity. To recognize the first description by Frank, et al. of their patient as a separate entity it was proposed to rename the disorder as Frank-ter Haar syndrome. Other patients have been reported previously as having the same condition [4-6]. Three probable cases were described as having the serpentine fibula syndrome (SFS) [7], another three possible patients were reported in an abstract [8], as was a pair of sibs with similar findings but severe mental retardation [9].
The gene responsible for Frank-ter Haar syndrome is SH3PXD2B gene. This gene encods a protein which is required for podosome formation and is involved in cell adhesion and migration of numerous cell types. Here we describe an Iranian 6-year-old boy with this syndrome which was confirmed by a homozygous pathogenic mutation in the SH3PXD2B gene.
This patient was a 5 years 8 months old Iranian boy, the firstborn child of a healthy parents and the only child of a complex consanguineous married couple. He was referred because of Multiple Congenital Anomalies and Dysmorphy. He was product of a normal and un-eventful pregnancy, and normal vaginal delivery. Proband's BW was 3030 gm., H. 48 cm., and HC 32 cm. His measurements at present are 16 kg., 105, and 52 cm.
The cardinal features were: Prominent and bossing forehead, wide frontal, down slanted palpebral fissure, posterior rotated ears, short nose and anteverted nostril, prominent lip, hypodontia, gum hypertrophy, narrow palate, micrognathia, inverted nipples, ventricular septal defect, kyphoscolisis, joint stiffness brachycamptodactyly in hands, flat feet, and undescended testis.
The diameter of left cornea was 14 mm. and the other 12 mm., and optic swelling (papilledema) with optic atrophy. On a recent cardiac Echo, the findings were: Thick and mixomatous mitral valve, floppy mitral and tricuspid valves, and a history of small VSD that was closed spontaneously.
I have searched the literature and relevant softwares, and found some diagnosis. The diagnoses were: Oral-Facial-Digital syn., Oculo-Dento-Digital syn., Oto-Palato-Digital syn. and Weaver syndrome. I myself think the last one is better match with the problem. There was a suggestion for CDGs too.
I contacted with Prof. Hennekam in the AMC Amsterdam and sent the complete phenotype of this case. I attached some of photos that may show the phenotype better and I asked him to take a look and let me have his Idea regarding this case? His suggestion after a long time correspondence was Frank-ter Haar syndrome, and referred me to Prof. Hans van Bokhoven in Nijmegen asking him whether he would be prepared to study this patient on a research basis for the known gene. Prof. van Bokhoven told me the entity is heterogeneous. He has a few families in which he has been unable to find a SH3PXD2B mutation. He did whole exome sequencing in them quite some time ago and has had a small number of candidate genes. He suggested that one of the families was from Pakistan and told that he is not sure from which region in Iran your family is coming from and are they relatives? Indeed this family was not relative of Pakistani family.
I sent the blood sample of the patient to Prof. Hans van Bokhoven in Nijmegen with the diagnosis of Frank-ter Haar syndrome. He analysed the SH3PXD2B gene and the result was a homozygous pathogenic mutation in the SH3PXD2B gene. It was a nonsense mutation c.115G > T(p.(Glu39*)) and truncation of the responsible protein and our clinical impression has been confirmed.
We present here an Iranian patient with clinical findings of Frank-ter-Haar syndrome. The combination of the craniofacial, cardiovascular, and skeletal anomalies support the unique nature of this syndrome. Comparison of the manifestation of this patient with those in previous reported is provided in the following table. Along with the symptoms regarded in the table there are additional signs such as downslanted palpebral fissure, posterior rotated ears, short nose and anteverted nostril, prominent lip, hypodontia, gum hypertrophy, narrow palate, micrognathia, inverted nipples, joint stiffness, brachycamptodactyly in hands, flat feet, and undescending testis [1-7].
Because of insufficient data the three cases described by Temtamy, et al. [8] were not included. No specific ophthalmological investigation has been performed but the local pediatrician stated the corneae to be unusually large.
Frank-ter Haar syndrome is a rare genetic disorder that results in brachycephaly, a large anterior fontanelle, a prominent forehead, hypertelorism, prominent eyes, megalocornea with or without glaucoma, full cheeks and flexion deformities at the Figure 1 and Figure 2. All of the above mentioned features of the syndrome were also observed in our case. We believe our patient's fontanelle was open with also stenosis of some sutures.
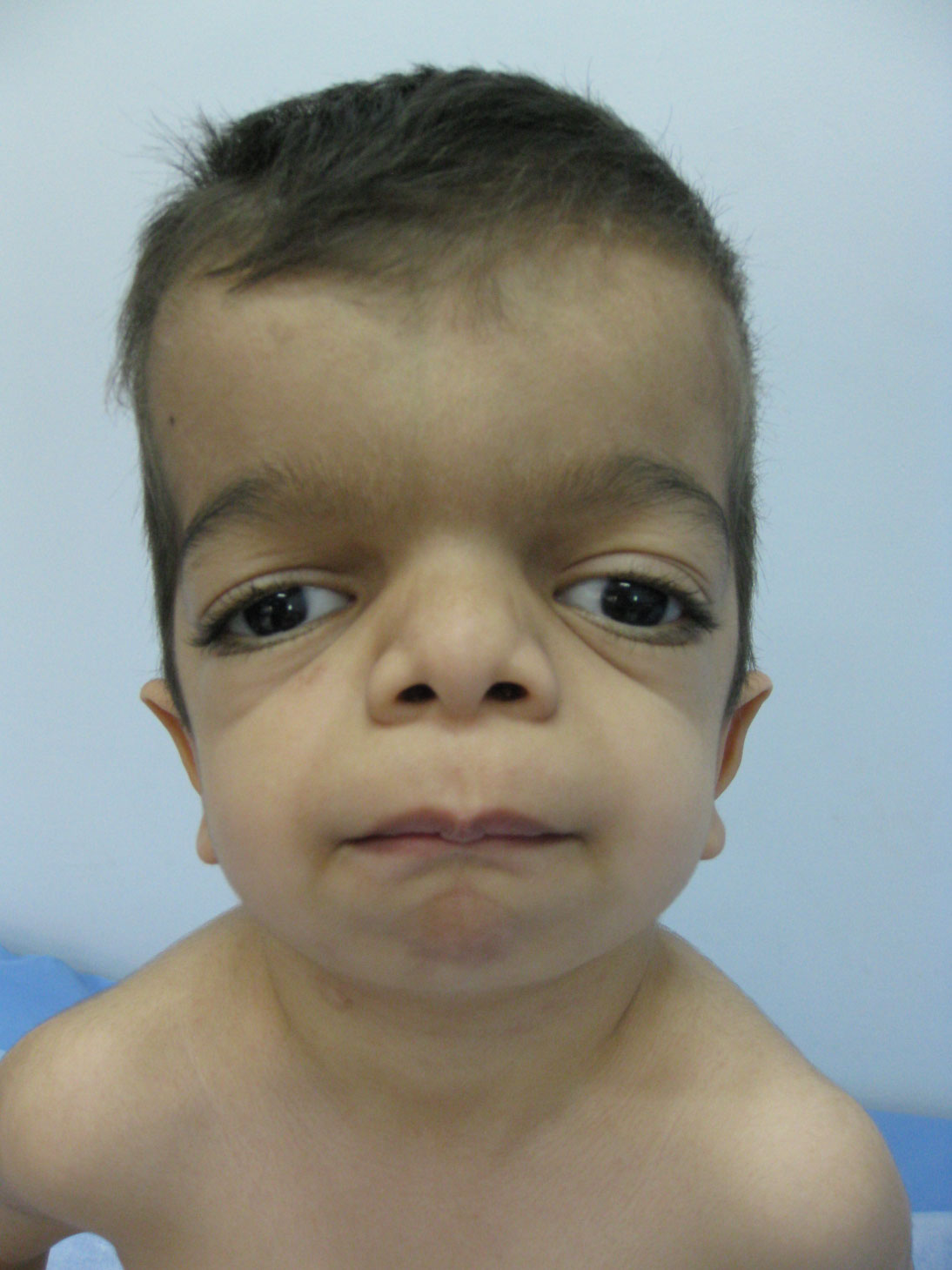 Figure 1: Facial Dysmorfisms: Full cheeks - Micrognathia - Course face; large eyes - Broad nasal tip - Anteverted nostrils; Malocclusion.
View Figure 1
Figure 1: Facial Dysmorfisms: Full cheeks - Micrognathia - Course face; large eyes - Broad nasal tip - Anteverted nostrils; Malocclusion.
View Figure 1
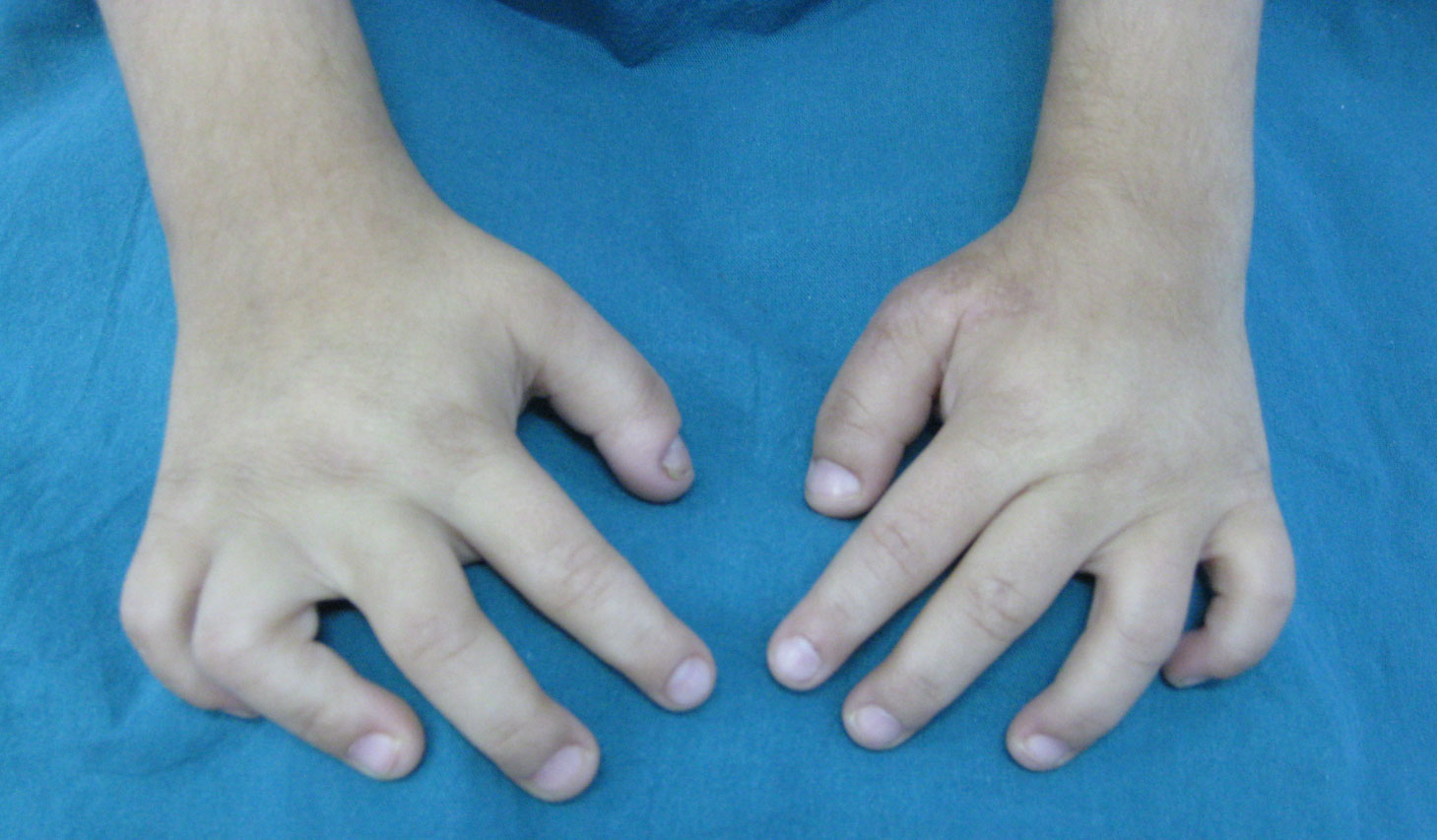 Figure 2: Flexion deformities of fingers - Short phalanges.
View Figure 2
Figure 2: Flexion deformities of fingers - Short phalanges.
View Figure 2
The findings in our case report are similar to the ones described earlier on Frank-ter Haar syndrome (Table 1), broad mouth, broad alveolar ridges, micrognathia, delayed dental development, delayed eruption, impacted teeth, anterior open bite, hypoplasia of the teeth, full cheeks, gingival hyperplasia and deep palate (Figure 3).
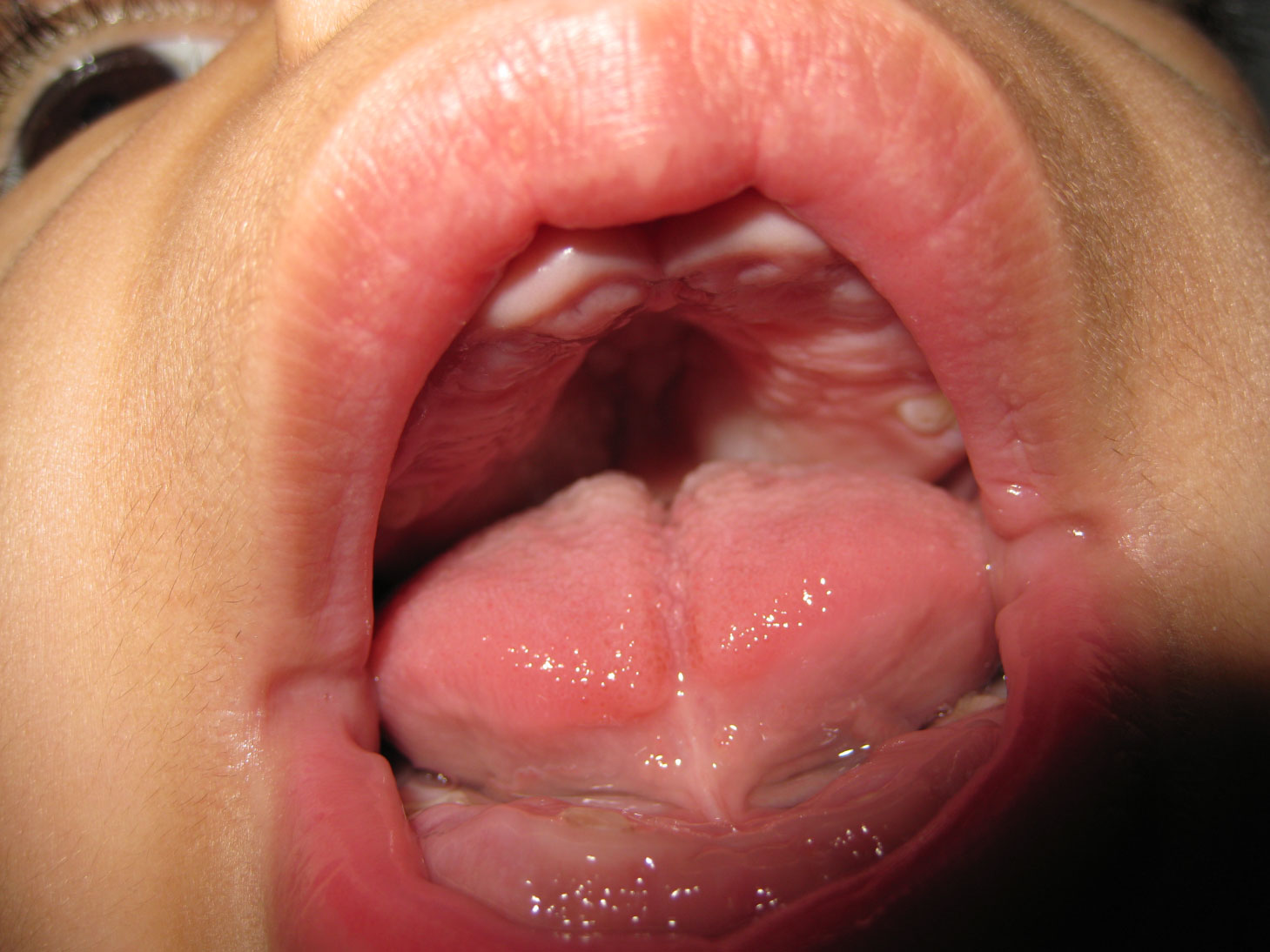 Figure 3: Mouth Deformities; High-arched palate - Gingival hypertrophy - Wide mouth.
View Figure 3
Figure 3: Mouth Deformities; High-arched palate - Gingival hypertrophy - Wide mouth.
View Figure 3
Table 1: Frank-ter Haar Syndrome. Comparison of the present patient with those from literature. View Table 1
Sinus underdevelopment or agenesis affects mainly the frontal sinuses; sphenoid sinus involvement is extremely rare [10]. Besides that, it is mentioned in the literature that, sinus agenesis or hypoplasia is seen in syndromes of craniosynostosis, osteodysplasia (Melnick-Needles syndrome) and Down syndrome [11] (Figure 4 and Figure 5). Our patient had agenesis of both the sphenoid and frontal sinuses and underdeveloped ethmoid and maxillary sinuses. There is no information about the paranasal sinuses of patients with Frank-ter Haar syndrome in the literature.
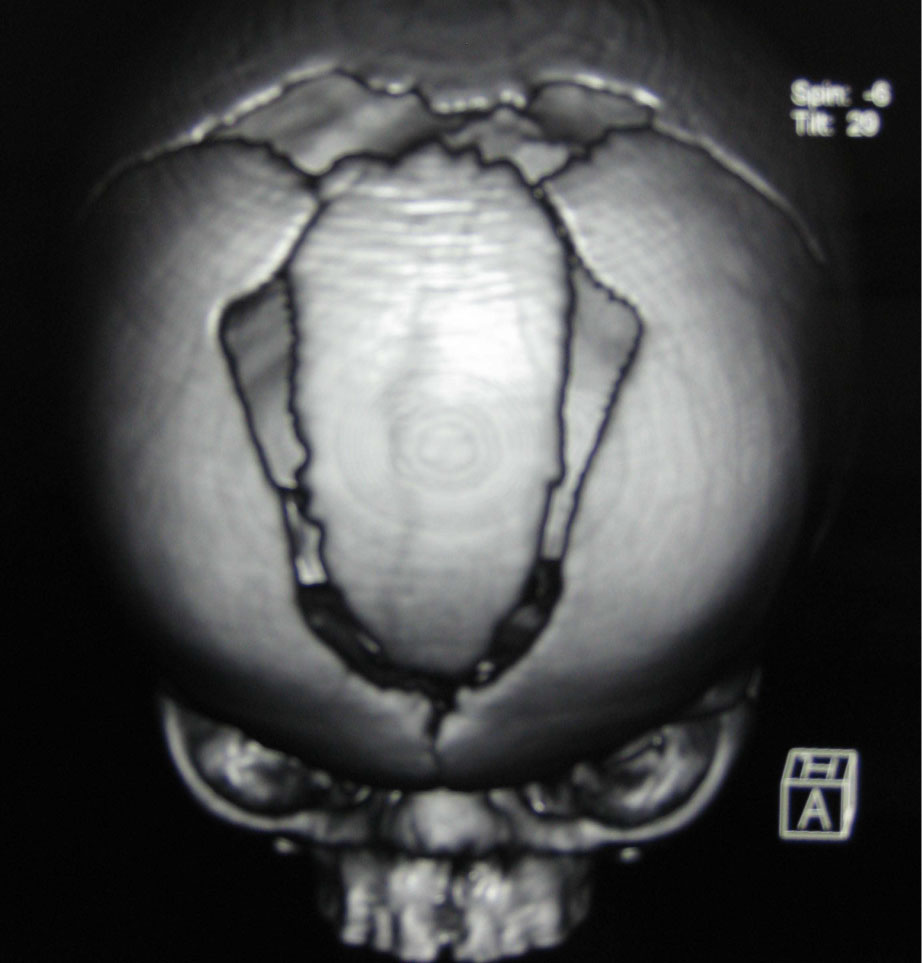 Figure 4: Sagittal craniosynostosis premature fusion of the calvarial sutures, skull MRI.
View Figure 4
Figure 4: Sagittal craniosynostosis premature fusion of the calvarial sutures, skull MRI.
View Figure 4
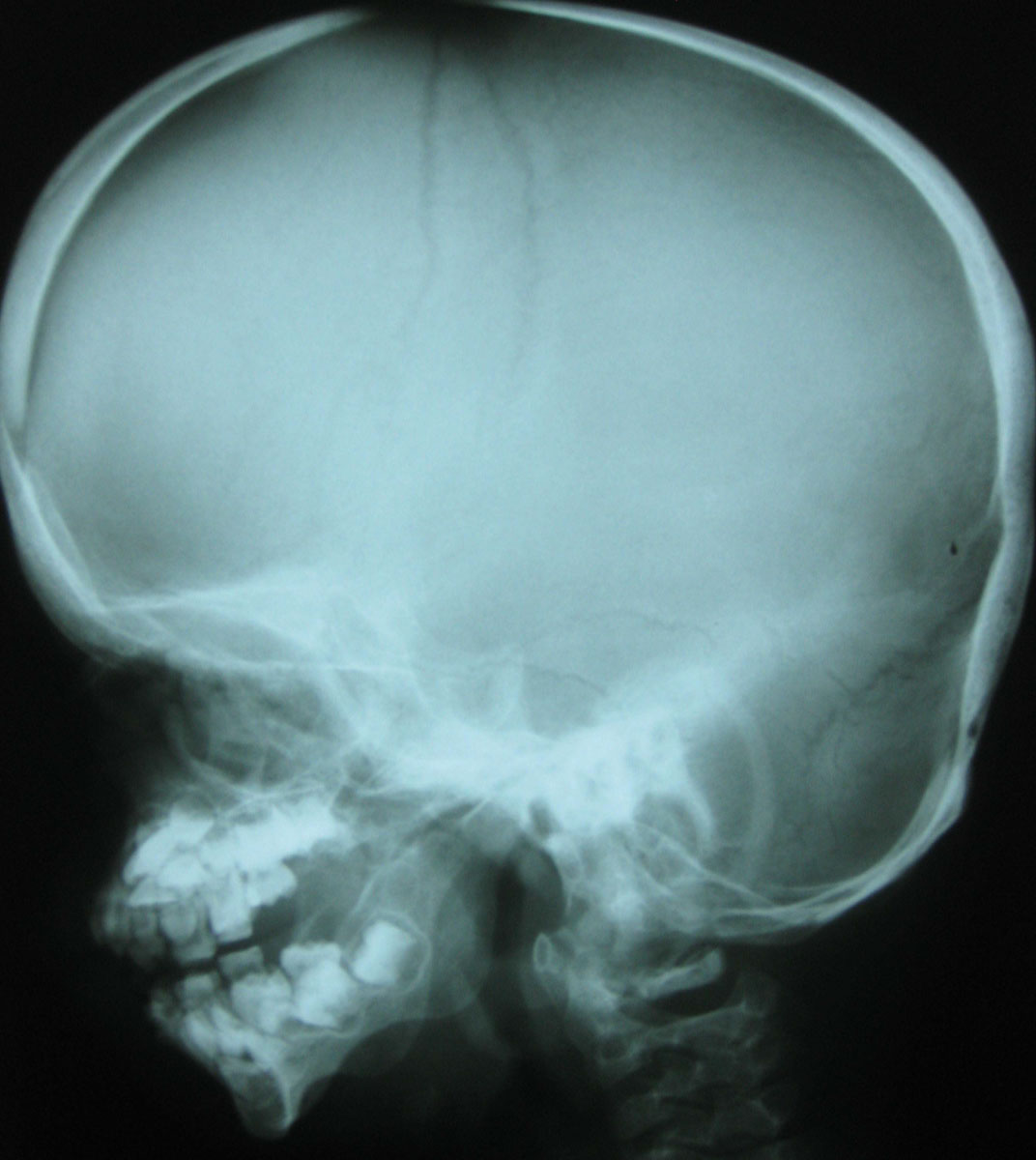 Figure 5: Skull X-Ray: Prominent forehead; small chin; brachycephaly; Wormian bones.
View Figure 5
Figure 5: Skull X-Ray: Prominent forehead; small chin; brachycephaly; Wormian bones.
View Figure 5
In conclusion, Frank-ter Haar syndrome is a rare autosomal recessive syndrome that involves various multiple maxillofacial anomalies. The report of our case outlines some of the anomalies seen in the patient with Frank-ter Haar syndrome.
Authors like to thank the family for co-operation in this study, and appreciate Prof. R. Hennekam for his kind collaboration in clinical evaluation of the case, and cordially thanks to Prof. H. van Bokhoven for his task for molecular analysis of this patient.